
Page 434 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 434
Chapter 29 Inherited Bone Marrow Failure Syndromes 355
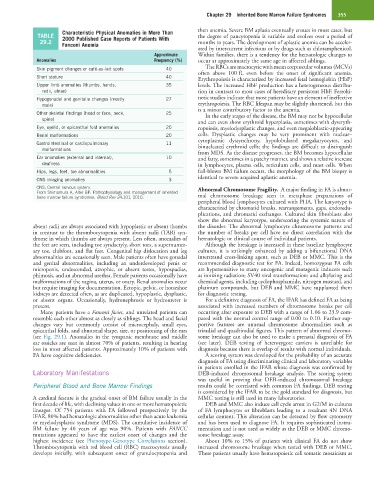
Characteristic Physical Anomalies in More Than then anemia. Severe BM aplasia eventually ensues in most cases, but
TABLE 2000 Published Case Reports of Patients With the degree of pancytopenia is variable and evolves over a period of
29.2 Fanconi Anemia months to years. The development of aplastic anemia can be acceler-
ated by intercurrent infections or by drugs such as chloramphenicol.
Approximate Within families, there is a tendency for the hematologic changes to
Anomalies Frequency (%) occur at approximately the same age in affected siblings.
Skin pigment changes or café-au-lait spots 40 The RBCs are macrocytic with mean corpuscular volumes (MCVs)
often above 100 fL even before the onset of significant anemia.
Short stature 40 Erythropoiesis is characterized by increased fetal hemoglobin (HbF)
Upper limb anomalies (thumbs, hands, 35 levels. The increased HbF production has a heterogeneous distribu-
radii, ulnae) tion in contrast to most cases of hereditary persistent HbF. Ferroki-
Hypogonadal and genitalia changes (mostly 27 netic studies indicate that most patients have an element of ineffective
male) erythropoiesis. The RBC lifespan may be slightly shortened, but this
is a minor contributory factor to the anemia.
Other skeletal findings (head or face, neck, 25 In the early stages of the disease, the BM may not be hypocellular
spine)
and can even show erythroid hyperplasia, sometimes with dyseryth-
Eye, eyelid, or epicanthal fold anomalies 20 ropoiesis, myelodysplastic changes, and even megaloblastic-appearing
Renal malformations 20 cells. Dysplastic changes may be very prominent with nuclear–
cytoplasmic dyssynchrony, hypolobulated megakaryocytes, and
Gastrointestinal or cardiopulmonary 11 binucleated erythroid cells; the findings are difficult to distinguish
malformations
from MDS. As the disease progresses, the BM becomes hypocellular
Ear anomalies (external and internal), 10 and fatty, sometimes in a patchy manner, and shows a relative increase
deafness in lymphocytes, plasma cells, reticulum cells, and mast cells. When
Hips, legs, feet, toe abnormalities 5 full-blown BM failure occurs, the morphology of the BM biopsy is
CNS imaging anomalies 3 identical to severe acquired aplastic anemia.
CNS, Central nervous system. Abnormal Chromosome Fragility. A major finding in FA is abnor-
From Shimamura A, Alter BP: Pathophysiology and management of inherited
bone marrow failure syndromes. Blood Rev 24:101, 2010. mal chromosome breakage seen in metaphase preparations of
peripheral blood lymphocytes cultured with PHA. The karyotype is
characterized by chromatid breaks, rearrangements, gaps, endoredu-
plications, and chromatid exchanges. Cultured skin fibroblasts also
show the abnormal karyotype, underscoring the systemic nature of
absent radii are always associated with hypoplastic or absent thumbs the disorder. The abnormal lymphocyte chromosome patterns and
in contrast to the thrombocytopenia with absent radii (TAR) syn- the number of breaks per cell have no direct correlation with the
drome in which thumbs are always present. Less often, anomalies of hematologic or clinical course of individual patients.
the feet are seen, including toe syndactyly, short toes, a supernumer- Although the breakage is increased in these baseline lymphocyte
ary toe, clubfoot, and flat feet. Congenital hip dislocation and leg cultures, it is strikingly enhanced by adding a bifunctional DNA
abnormalities are occasionally seen. Male patients often have gonadal interstrand cross-linking agent, such as DEB or MMC. This is the
and genital abnormalities, including an underdeveloped penis or recommended diagnostic test for FA. Indeed, homozygous FA cells
micropenis, undescended, atrophic, or absent testes, hypospadias, are hypersensitive to many oncogenic and mutagenic inducers such
phimosis, and an abnormal urethra. Female patients occasionally have as ionizing radiation; SV40 viral transformation; and alkylating and
malformations of the vagina, uterus, or ovary. Renal anomalies occur chemical agents, including cyclophosphamide, nitrogen mustard, and
but require imaging for documentation. Ectopic, pelvic, or horseshoe platinum compounds, but DEB and MMC have supplanted them
kidneys are detected often, as are duplicated, hypoplastic, dysplastic, for diagnostic testing.
or absent organs. Occasionally, hydronephrosis or hydroureter is For a definitive diagnosis of FA, the IFAR has defined FA as being
present. associated with increased numbers of chromosome breaks per cell
Many patients have a Fanconi facies, and unrelated patients can occurring after exposure to DEB with a range of 1.06 to 23.9 com-
resemble each other almost as closely as siblings. The head and facial pared with the normal control range of 0.00 to 0.10. Further sup-
changes vary but commonly consist of microcephaly, small eyes, portive features are unusual chromosome abnormalities such as
epicanthal folds, and abnormal shape, size, or positioning of the ears triradial and quadriradial figures. This pattern of abnormal chromo-
(see Fig. 29.1). Anomalies in the tympanic membrane and middle some breakage can also be used to make a prenatal diagnosis of FA
ear ossicles are seen in almost 70% of patients, resulting in hearing (see later). DEB testing of heterozygote carriers is unreliable for
loss in most affected patients. Approximately 10% of patients with diagnosis because there is overlap of results with normal individuals.
FA have cognitive deficiencies. A scoring system was developed for the probability of an accurate
diagnosis of FA using discriminating clinical and laboratory variables
in patients enrolled in the IFAR whose diagnosis was confirmed by
Laboratory Manifestations DEB-induced chromosomal breakage analysis. The scoring system
was useful in proving that DEB-induced chromosomal breakage
Peripheral Blood and Bone Marrow Findings results could be correlated with common FA findings. DEB testing
is considered by the IFAR to be the gold standard for diagnosis, but
A cardinal feature is the gradual onset of BM failure usually in the MMC testing is still used in many laboratories.
first decade of life, with declining values in one or more hematopoietic DEB and MMC also induce cell cycle arrest in G2/M in cultures
lineages. Of 754 patients with FA followed prospectively by the of FA lymphocytes or fibroblasts leading to a resultant 4N DNA
IFAR, 80% had hematologic abnormalities other than acute leukemia cellular content. This alteration can be detected by flow cytometry
or myelodysplastic syndrome (MDS). The cumulative incidence of and has been used to diagnose FA. It requires sophisticated instru-
BM failure by 40 years of age was 90%. Patients with FANCC mentation and is not used as widely as the DEB or MMC chromo-
mutations appeared to have the earliest onset of changes and the some breakage assay.
highest incidence (see Phenotype-Genotype Correlations section). About 10% to 15% of patients with clinical FA do not show
Thrombocytopenia with red blood cell (RBC) macrocytosis usually increased chromosome breakage when tested with DEB or MMC.
develops initially, with subsequent onset of granulocytopenia and These patients usually have hematopoietic cell somatic mosaicism as