
Page 1238 - Williams Hematology ( PDFDrive )
P. 1238
1212 Part IX: Lymphocytes and Plasma Cells Chapter 80: Immunodeficiency Diseases 1213
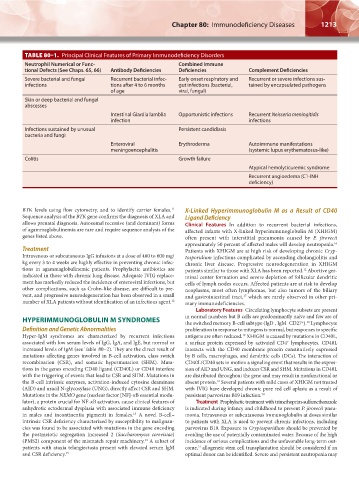
TABLE 80–1. Principal Clinical Features of Primary Immunodeficiency Disorders
Neutrophil Numerical or Func- Combined Immune
tional Defects (See Chaps. 65, 66) Antibody Deficiencies Deficiencies Complement Deficiencies
Severe bacterial and fungal Recurrent bacterial infec- Early onset respiratory and Recurrent or severe infections sus-
infections tions after 4 to 6 months gut infections (bacterial, tained by encapsulated pathogens
of age viral, fungal)
Skin or deep bacterial and fungal
abscesses
Intestinal Giardia lamblia Opportunistic infections Recurrent Neisseria meningitidis
infection infections
Infections sustained by unusual Persistent candidiasis
bacteria and fungi
Enteroviral Erythroderma Autoimmune manifestations
meningoencephalitis (systemic lupus erythematosus-like)
Colitis Growth failure
Atypical hemolyticuremic syndrome
Recurrent angioedema (C1-INH
deficiency)
11
BTK levels using flow cytometry, and to identify carrier females. X-Linked Hyperimmunoglobulin M as a Result of CD40
Sequence analysis of the BTK gene confirms the diagnosis of XLA and Ligand Deficiency
allows prenatal diagnosis. Autosomal recessive (and dominant) forms Clinical Features In addition to recurrent bacterial infections,
of agammaglobulinemia are rare and require sequence analysis of the affected infants with X-linked hyperimmunoglobulin M (XHIGM)
genes listed above. often present with interstitial pneumonia caused by P. jirovecii
approximately 50 percent of affected males will develop neutropenia.
16
Treatment Patients with XHIGM are at high risk of developing chronic Cryp-
Intravenous or subcutaneous IgG infusions at a dose of 400 to 600 mg/ tosporidium infections complicated by ascending cholangiolitis and
kg every 3 to 4 weeks are highly effective in preventing chronic infec- chronic liver disease. Progressive neurodegeneration in XHIGM
tions in agammaglobulinemic patients. Prophylactic antibiotics are patients similar to those with XLA has been reported. Abortive ger-
12
indicated in those with chronic lung disease. Adequate IVIG replace- minal center formation and severe depletion of follicular dendritic
ment has markedly reduced the incidence of enteroviral infections, but cells of lymph nodes occurs. Affected patients are at risk to develop
other complications, such as Crohn-like disease, are difficult to pre- neoplasms, most often lymphomas, but also tumors of the biliary
vent, and progressive neurodegeneration has been observed in a small and gastrointestinal tract, which are rarely observed in other pri-
17
number of XLA patients without identification of an infectious agent. 12 mary immunodeficiencies.
Laboratory Features Circulating lymphocyte subsets are present
HYPERIMMUNOGLOBULIN M SYNDROMES in normal numbers but B cells are predominantly naïve and few are of
the switched memory B-cell subtype (IgD , IgM CD27 ). Lymphocyte
+ 18
–
–
Definition and Genetic Abnormalities proliferation in response to mitogens is normal, but responses to specific
Hyper-IgM syndromes are characterized by recurrent infections antigens are often reduced. XHIGM is caused by mutations in CD40L,
19
associated with low serum levels of IgG, IgA, and IgE, but normal or a surface protein expressed by activated CD4 lymphocytes. CD40L
+
increased levels of IgM (see Table 80–2). They are the direct result of interacts with the CD40 membrane protein constitutively expressed
mutations affecting genes involved in B-cell activation, class switch by B cells, macrophages, and dendritic cells (DCs). The interaction of
recombination (CSR), and somatic hypermutation (SHM). Muta- CD40L/CD40 sets in motion a signaling event that results in the expres-
tions in the genes encoding CD40 ligand (CD40L) or CD40 interfere sion of AID and UNG, and induces CSR and SHM. Mutations in CD40L
with the triggering of events that lead to CSR and SHM. Mutations in are distributed throughout the gene and may result in nonfunctional or
the B-cell intrinsic enzymes, activation-induced cytosine deaminase absent protein. Several patients with mild cases of XHIGM not treated
16
(AID) and uracil N-glycosylase (UNG), directly affect CSR and SHM. with IVIG have developed chronic pure red cell aplasia as a result of
Mutations in the NEMO gene (nuclear factor [NF]-κB essential modu- persistent parvovirus B19 infection. 20
lator), a protein crucial for NF-κB activation, cause clinical features of Treatment Prophylactic treatment with trimethoprim-sulfamethoxazole
anhydrotic ectodermal dysplasia with associated immune deficiency is indicated during infancy and childhood to prevent P. jirovecii pneu-
in males and incontinentia pigmenti in females. A novel B-cell– monia. Intravenous or subcutaneous immunoglobulin at doses similar
13
intrinsic CSR deficiency characterized by susceptibility to malignan- to patients with XLA is used to prevent chronic infections, including
cies was found to be associated with mutations in the gene encoding parvovirus B19. Exposure to Cryptosporidium should be prevented by
the postmeiotic segregation increased 2 (Saccharomyces cerevisiae) avoiding the use of potentially contaminated water. Because of the high
(PMS2) component of the mismatch repair machinery. A subset of incidence of serious complications and the unfavorable long-term out-
14
patients with ataxia telangiectasia present with elevated serum IgM come, allogeneic stem cell transplantation should be considered if an
21
and CSR deficiency. 15 optimal donor can be identified. Severe and persistent neutropenia may
Kaushansky_chapter 80_p1211-1238.indd 1213 9/18/15 10:00 AM