
Page 942 - Williams Hematology ( PDFDrive )
P. 942
916 Part VI: The Erythrocyte Chapter 59: Polyclonal and Hereditary Sideroblastic Anemias 917
Diferric
transferrin
Apo-
Cell
transferrin membrane
Transferrin
receptor
STEAP 3
NAD(P)H Clathrin-coated pit
NAD(P)
DMT 1
Endocytosis
Exocytosis
+
H proton pump
Hgb globin + heme
STEAP 3
Copro’gen
?
?
Proto’gen Outer membrane
?
CPO PPO Mitoferrin 1
FECH Inner membrane
Proto IX
Heme
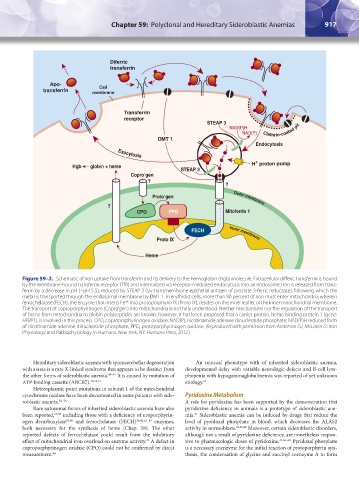
Figure 59–2. Schematic of iron uptake from transferrin and its delivery to the hemoglobin (Hgb) molecule. Extracellular differic transferrin is bound
by the membrane-bound transferrin receptor (TfR) and internalized via receptor-mediated endocytosis into an endosome. Iron is released from trans-
ferrin by a decrease in pH (~pH 5.5), reduced by STEAP 3 (six-transmembrane epithelial antigen of prostate 3-ferric reductase), following which the
metal is transported through the endosomal membrane by DMT 1. In erythroid cells, more than 90 percent of iron must enter mitochondria wherein
ferrochelatase (FECH), the enzyme that inserts Fe into protoporphyrin IX (Proto IX), resides on the inner leaflet of the inner mitochondrial membrane.
2+
The transport of coproporphyrinogen (Copro’gen) into mitochondria is not fully understood. Neither mechanisms nor the regulation of the transport
of heme from mitochondria to globin polypeptides are known; however, it has been proposed that a carrier protein, heme binding protein 1 (gene:
HEBP1), is involved in this process. CPO, coproporphyrinogen oxidase; NAD(P), nicotinamide adenine dinucleotide phosphate; NAD(P)H reduced form
of nicotinamide adenine dinucleotide phosphate; PPO, protoporphyrinogen oxidase. (Reproduced with permission from Anderson GJ, McLaren G: Iron
Physiology and Pathophysiology in Humans. New York, NY: Humana Press; 2012.)
Hereditary sideroblastic anemia with spinocerebellar degeneration An unusual phenotype with of inherited sideroblastic anemia,
with ataxia is a rare X-linked syndrome that appears to be distinct from developmental delay with variable neurologic defects and B-cell lym-
the other forms of sideroblastic anemia. 48–51 It is caused by mutation of phopenia with hypogammaglobulinemia was reported of yet unknown
ATP-binding cassette (ABCB7). 48,52,53 etiology. 65
Heteroplasmic point mutations in subunit 1 of the mitochondrial
cytochrome oxidase have been documented in some patients with side- Pyridoxine Metabolism
roblastic anemia. 54–56 A role for pyridoxine has been supported by the demonstration that
Rare autosomal forms of inherited sideroblastic anemia have also pyridoxine deficiency in animals is a prototype of sideroblastic ane-
been reported, 57,58 including those with a deficiency of uroporphyrin- mia. Sideroblastic anemia can be induced by drugs that reduce the
31
ogen decarboxylase 59,60 and ferrochelatase (FECH) 36,41,61–63 enzymes, level of pyridoxal phosphate in blood, which decreases the ALAS2
both necessary for the synthesis of heme (Chap. 58). The other activity in normoblasts. 22,36,40 Moreover, certain sideroblastic disorders,
reported defects of ferrochelatase could result from the inhibitory although not a result of pyridoxine deficiency, are nonetheless respon-
effect of mitochondrial iron overload on enzyme activity. A defect in sive to pharmacologic doses of pyridoxine. 44,66–68 Pyridoxal phosphate
41
coproporphyrinogen oxidase (CPO) could not be confirmed by direct is a necessary coenzyme for the initial reaction of protoporphyrin syn-
measurement. 64 thesis, the condensation of glycine and succinyl coenzyme A to form
Kaushansky_chapter 59_p0915-0922.indd 917 9/17/15 3:17 PM