
Page 1033 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 1033
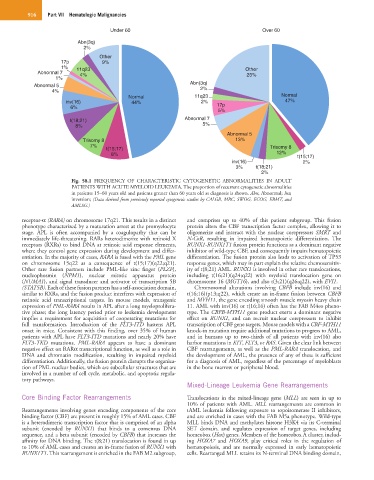
916 Part VII Hematologic Malignancies
Under 60 Over 60
Abn(3q)
2%
Other
17p 9%
1% 11q23 Other
Abnormal 7 4% 25%
1%
Abn(3q)
Abnormal 5
4% 2%
Normal 11q23 Normal
inv(16) 44% 2% 47%
6% 17p
5%
t(18;21) Abnormal 7
8% 5%
Abnormal 5
Trisomy 8 13%
7% Trisomy 8
t(15;17)
8% 12% t(15;17)
inv(16) 2%
3% t(18;21)
2%
Fig. 58.1 FREQUENCY OF CHARACTERISTIC CYTOGENETIC ABNORMALITIES IN ADULT
PATIENTS WITH ACUTE MYELOID LEUKEMIA. The proportion of recurrent cytogenetic abnormalities
in patients 15–60 years old and patients greater than 60 years old at diagnosis is shown. Abn, Abnormal; Inv,
inversion; (Data derived from previously reported cytogenetic studies by CALGB, MRC, SWOG, ECOG, EBMT, and
AMLSG.)
receptor-α (RARA) on chromosome 17q21. This results in a distinct and comprises up to 40% of this patient subgroup. This fusion
phenotype characterized by a maturation arrest at the promyelocyte protein alters the CBF transcription factor complex, allowing it to
stage. APL is often accompanied by a coagulopathy that can be oligomerize and interact with the nuclear corepressors SMRT and
immediately life-threatening. RARs heterodimerize with retinoid X N-CoR, resulting in impaired hematopoietic differentiation. The
receptors (RXRs) to bind DNA at retinoic acid response elements, RUNX1-RUNX1T1 fusion protein functions as a dominant negative
where they control gene expression during development and differ- inhibitor of wild-type CBF, and consequently impairs hematopoietic
entiation. In the majority of cases, RARA is fused with the PML gene differentiation. The fusion protein also leads to activation of TP53
on chromosome 15q22 as a consequence of t(15;17)(q22;q21). response genes, which may in part explain the relative chemosensitiv-
Other rare fusion partners include PML-like zinc finger (PLZF), ity of t(8;21) AML. RUNX1 is involved in other rare translocations,
nucleophosmin (NPM1), nuclear mitotic apparatus protein including t(16;21)(q24;q22) with myeloid translocation gene on
(NUMA1), and signal transducer and activator of transcription 5B chromosome 16 (MGT16), and also t(3;21)(q26;q22), with EVI1.
(STAT5B). Each of these fusion partners has a self-association domain, Chromosomal alterations involving CBFB include inv(16) and
similar to RXRs, and the fusion product interferes with expression of t(16;16)(p13;q22), which create an in-frame fusion between CBFB
retinoic acid transcriptional targets. In mouse models, transgenic and MYH11, the gene encoding smooth muscle myosin heavy chain
expression of PML-RARA results in APL after a long myeloprolifera- 11. AML with inv(16) or t(16;16) often has the FAB M4eo pheno-
tive phase; the long latency period prior to leukemia development type. The CBFB-MYH11 gene product exerts a dominant negative
implies a requirement for acquisition of cooperating mutations for effect on RUNX1, and can recruit nuclear corepressors to inhibit
full transformation. Introduction of the FLT3-ITD hastens APL transcription of CBF gene targets. Mouse models with a CBF-MYH11
onset in mice. Consistent with this finding, over 35% of human knock-in mutation require additional mutations to progress to AML,
patients with APL have FLT3-ITD mutations and nearly 20% have and in humans up to two-thirds of all patients with inv(16) also
FLT3-TKD mutations. PML-RARA appears to have a dominant harbor mutations in KIT, FLT3, or RAS. Given the clear link between
negative effect on RARα transcriptional function, as well as a role in CBF rearrangements, as well as the PML-RARA translocation, and
DNA and chromatin modification, resulting in impaired myeloid the development of AML, the presence of any of these is sufficient
differentiation. Additionally, the fusion protein disrupts the organiza- for a diagnosis of AML, regardless of the percentage of myeloblasts
tion of PML nuclear bodies, which are subcellular structures that are in the bone marrow or peripheral blood.
involved in a number of cell cycle, metabolic, and apoptotic regula-
tory pathways.
Mixed-Lineage Leukemia Gene Rearrangements
Core Binding Factor Rearrangements Translocations in the mixed-lineage gene (MLL) are seen in up to
10% of patients with AML. MLL rearrangements are common in
Rearrangements involving genes encoding components of the core tAML leukemia following exposure to topoisomerase II inhibitors,
binding factor (CBF) are present in roughly 15% of AML cases. CBF and are enriched in cases with the FAB M5a phenotype. Wild-type
is a heterodimeric transcription factor that is comprised of an alpha MLL binds DNA and methylates histone H3K4 via its C-terminal
subunit (encoded by RUNX1) that binds to a consensus DNA SET domain, and regulates expression of target genes, including
sequence, and a beta subunit (encoded by CBFB) that increases the homeobox (Hox) genes. Members of the homeobox A cluster, includ-
affinity for DNA binding. The t(8;21) translocation is found in up ing HOXA7 and HOXA9, play critical roles in the regulation of
to 10% of AML cases and creates an in-frame fusion of RUNX1 with hematopoiesis, and are normally expressed in early hematopoietic
RUNX1T1. This rearrangement is enriched in the FAB M2 subgroup, cells. Rearranged MLL retains its N-terminal DNA binding domain,