
Page 1035 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 1035
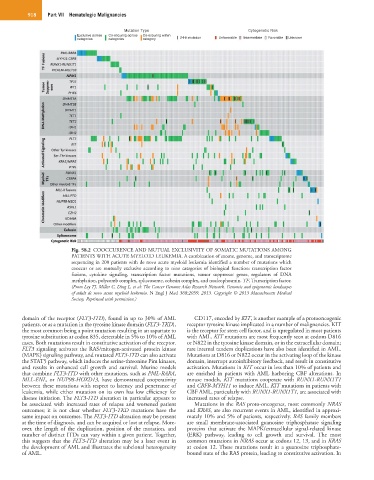
918 Part VII Hematologic Malignancies
Mutation Type Cytogenetic Risk
Exclusive across Co-occuring across Co-occuring within
categories categories category 2-Hit mutation Unfavorable Intermediate Favorable Unknown
Fig. 58.2 COOCCURENCE AND MUTUAL EXCLUSIVITY OF SOMATIC MUTATIONS AMONG
PATIENTS WITH ACUTE MYELOID LEUKEMIA. A combination of exome, genome, and transcriptome
sequencing in 200 patients with de novo acute myeloid leukemia identified a number of mutations which
cooccur or are mutually exclusive according to nine categories of biological function: transcription factor
fusions, cytokine signaling, transcription factor mutations, tumor suppressor genes, regulators of DNA
methylation, polycomb complex, spliceosome, cohesin complex, and nucleophosmin. TF, Transcription factor.
(From Ley TJ, Miller C, Ding L, et al: The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes
of adult de novo acute myeloid leukemia. N Engl J Med 368:2059, 2013. Copyright © 2013 Massachusetts Medical
Society. Reprinted with permission.)
domain of the receptor (FLT3-ITD), found in up to 30% of AML CD117, encoded by KIT, is another example of a protooncogenic
patients, or as a mutation in the tyrosine kinase domain (FLT3-TKD), receptor tyrosine kinase implicated in a number of malignancies. KIT
the most common being a point mutation resulting in an aspartate to is the receptor for stem cell factor, and is upregulated in most patients
tyrosine substitution at codon 835, detectable in 5% to 10% of AML with AML. KIT mutations are most frequently seen at codons D816
cases. Both mutations result in constitutive activation of the receptor. or N822 in the tyrosine kinase domain, or in the extracellular domain;
FLT3 signaling activates the RAS/mitogen-activated protein kinase rare internal tandem duplications have also been identified in AML.
(MAPK) signaling pathway, and mutated FLT3-ITD can also activate Mutations at D816 or N822 occur in the activating loop of the kinase
the STAT5 pathway, which induces the serine-threonine Pim kinases, domain, interrupt autoinhibitory feedback, and result in constitutive
and results in enhanced cell growth and survival. Murine models activation. Mutations in KIT occur in less than 10% of patients and
that combine FLT3-ITD with other mutations, such as PML-RARA, are enriched in patients with AML harboring CBF alterations. In
MLL-ENL, or NUP98-HOXD13, have demonstrated cooperativity mouse models, KIT mutations cooperate with RUNX1-RUNX1T1
between these mutations with respect to latency and penetrance of and CBFB-MYH11 to induce AML. KIT mutations in patients with
leukemia, while either mutation on its own has low efficiency for CBF AML, particularly with RUNX1-RUNX1T1, are associated with
disease initiation. The FLT3-ITD alteration in particular appears to increased rates of relapse.
be associated with increased rates of relapse and worsened patient Mutations in the RAS proto-oncogenes, most commonly NRAS
outcomes; it is not clear whether FLT3-TKD mutations have the and KRAS, are also recurrent events in AML, identified in approxi-
same impact on outcomes. The FLT3-ITD alteration may be present mately 10% and 5% of patients, respectively. RAS family members
at the time of diagnosis, and can be acquired or lost at relapse. More- are small membrane-associated guanosine triphosphatase signaling
over, the length of the duplication, position of the mutation, and proteins that activate the MAPK/extracellular signal-related kinase
number of distinct ITDs can vary within a given patient. Together, (ERK) pathway, leading to cell growth and survival. The most
this suggests that the FLT3-ITD alteration may be a later event in common mutations in NRAS occur at codons 12, 13, and in KRAS
the development of AML and illustrates the subclonal heterogeneity at codon 12. These mutations result in a guanosine triphosphate-
of AML. bound state of the RAS protein, leading to constitutive activation. In