
Page 2003 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 2003
Chapter 117 Transfusion Therapy for Coagulation Factor Deficiencies 1777
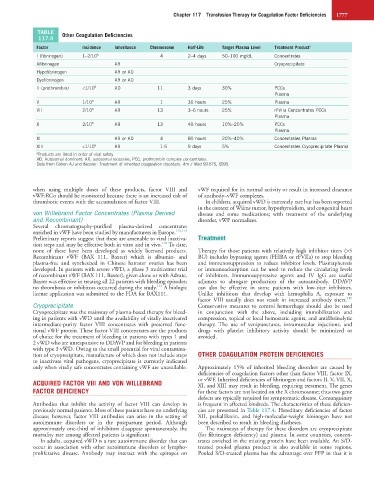
TABLE Other Coagulation Deficiencies
117.4
Factor Incidence Inheritance Chromosome Half-Life Target Plasma Level Treatment Product a
I (fibrinogen) 1–2/10 6 4 2–4 days 50–100 mg/dL Concentrates
Afibrinogen AR Cryoprecipitate
Hypofibrinogen AR or AD
Dysfibrinogen AR or AD
II (prothrombin) <1/10 6 AD 11 3 days 30% PCCs
Plasma
V 1/10 6 AR 1 36 hours 25% Plasma
VII 2/10 6 AR 13 3–6 hours 25% rFVIIa Concentrates PCCs
Plasma
X 2/10 6 AR 13 40 hours 10%–25% PCCs
Plasma
XI AR or AD 4 80 hours 20%–40% Concentrates Plasma
XIII <1/10 6 AR 1.6 9 days 5% Concentrates Cryoprecipitate Plasma
a Products are listed in order of viral safety.
AD, Autosomal dominant; AR, autosomal recessive; PCC, prothrombin complex concentrates.
Data from Cohen AJ and Kessler: Treatment of inherited coagulation disorders. Am J Med 99:675, 1995.
when using multiple doses of these products, factor VIII and vWF required for its normal activity or result in increased clearance
vWF:RCo should be monitored because there is an increased risk of of antibody–vWF complexes.
thrombotic events with the accumulation of factor VIII. In children, acquired vWD is extremely rare but has been reported
in the context of Wilms tumor, hypothyroidism, and congenital heart
von Willebrand Factor Concentrates (Plasma Derived disease and some medications; with treatment of the underlying
and Recombinant) disorder, vWF normalizes.
Several chromatography-purified plasma-derived concentrates
enriched in vWF have been studied by manufacturers in Europe. 112,113
Preliminary reports suggest that these are amenable to viral inactiva- Treatment
110
tion steps and may be effective both in vitro and in vivo. To date,
none of these have been developed as widely licensed products. Therapy for those patients with relatively high inhibitor titers (>5
Recombinant vWF (BAX 111, Baxter) which is albumin- and BU) includes bypassing agents (FEIBA or rFVIIa) to stop bleeding
plasma-free and synthesized in Chinese hamster ovaries has been and immunosuppression to reduce inhibitor levels. Plasmapheresis
developed. In patients with severe vWD, a phase 3 multicenter trial or immunoadsorption can be used to reduce the circulating levels
of recombinant vWF (BAX 111, Baxter), given alone or with Advate, of inhibitors. Immunosuppressive agents and IV IgG are useful
Baxter was effective in treating all 22 patients with bleeding episodes; adjuncts to abrogate production of the autoantibody. DDAVP
114
no thrombosis or inhibitors occurred during the study. A biologic can also be effective in some patients with low-titer inhibitors.
license application was submitted to the FDA for BAX111. Unlike inhibitors that develop with hemophilia A, exposure to
115
factor VIII usually does not result in increased antibody titers.
Cryoprecipitate Conservative measures to control hemorrhage should also be used
Cryoprecipitate was the mainstay of plasma-based therapy for bleed- in conjunction with the above, including immobilization and
ing in patients with vWD until the availability of virally inactivated compression, topical or local hemostatic agents, and antifibrinolytic
intermediate-purity factor VIII concentrates with preserved func- therapy. The use of venipunctures, intramuscular injections, and
tional vWF protein. These factor VIII concentrates are the products drugs with platelet inhibitory activity should be minimized or
of choice for the treatment of bleeding in patients with types 1 and avoided.
2 vWD who are unresponsive to DDAVP and for bleeding in patients
with type 3 vWD. Owing to the small potential for viral contamina-
tion of cryoprecipitate, manufacture of which does not include steps OTHER COAGULATION PROTEIN DEFICIENCIES
to inactivate viral pathogens, cryoprecipitate is currently indicated
only when virally safe concentrates containing vWF are unavailable. Approximately 15% of inherited bleeding disorders are caused by
deficiencies of coagulation factors other than factor VIII, factor IX,
ACQUIRED FACTOR VIII AND VON WILLEBRAND or vWF. Inherited deficiencies of fibrinogen and factors II, V, VII, X,
XI, and XIII may result in bleeding, requiring treatment. The genes
FACTOR DEFICIENCY for these factors are not located on the X chromosome; thus two gene
defects are typically required for symptomatic disease. Consanguinity
Antibodies that inhibit the activity of factor VIII can develop in is frequent in affected kindreds. The characteristics of these deficien-
previously normal patients. Most of these patients have no underlying cies are presented in Table 117.4. Hereditary deficiencies of factor
disease; however, factor VIII antibodies can arise in the setting of XII, prekallikrein, and high-molecular-weight kininogen have not
autoimmune disorders or in the postpartum period. Although been described to result in bleeding diatheses.
approximately one-third of inhibitors disappear spontaneously, the The mainstays of therapy for these disorders are cryoprecipitate
mortality rate among affected patients is significant. (for fibrinogen deficiency) and plasma. In some countries, concen-
In adults, acquired vWD is a rare autoimmune disorder that can trates enriched in the missing protein have been available. An S/D-
occur in association with other autoimmune disorders or lympho- treated pooled plasma product is also available in some regions.
proliferative disease. Antibody may interact with the epitopes on Pooled S/D-treated plasma has the advantage over FFP in that it is