
Page 448 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 448
Chapter 29 Inherited Bone Marrow Failure Syndromes 369
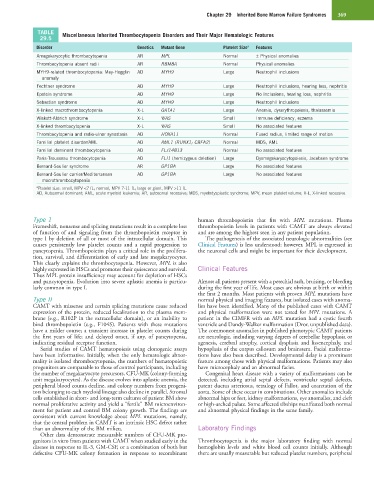
TABLE Miscellaneous Inherited Thrombocytopenia Disorders and Their Major Hematologic Features
29.5
Disorder Genetics Mutant Gene Platelet Size a Features
Amegakaryocytic thrombocytopenia AR MPL Normal ± Physical anomalies
Thrombocytopenia absent radii AR RBM8A Normal Physical anomalies
MYH9-related thrombocytopenia: May-Hegglin AD MYH9 Large Neutrophil inclusions
anomaly
Fechtner syndrome AD MYH9 Large Neutrophil inclusions, hearing loss, nephritis
Epstein syndrome AD MYH9 Large No inclusions, hearing loss, nephritis
Sebastian syndrome AD MYH9 Large Neutrophil inclusions
X-linked macrothrombocytopenia X-L GATA1 Large Anemia, dyserythropoiesis, thalassemia
Wiskott-Aldrich syndrome X-L WAS Small Immune deficiency, eczema
X-linked thrombocytopenia X-L WAS Small No associated features
Thrombocytopenia and radio-ulnar synostosis AD HOXA11 Normal Fused radius, limited range of motion
Familial platelet disorder/AML AD AML1 (RUNX1; CBFA2) Normal MDS, AML
Familial dominant thrombocytopenia AD FLJ14813 Normal No associated features
Paris-Trousseau thrombocytopenia AD FLI1 (hemizygous deletion) Large Dysmegakaryocytopoiesis, Jacobsen syndrome
Bernard-Soulier syndrome AR GP1BA Large No associated features
Bernard-Soulier carrier/Mediterranean AD GP1BA Large No associated features
macrothrombocytopenia
a Platelet size: small, MPV <7 fL; normal, MPV 7-11 fL; large or giant, MPV >11 fL.
AD, Autosomal dominant; AML, acute myeloid leukemia; AR, autosomal recessive; MDS, myelodysplastic syndrome; MPV, mean platelet volume; X-L, X-linked recessive.
Type 1 human thrombopoietin that fits with MPL mutations. Plasma
Frameshift, nonsense and splicing mutations result in a complete loss thrombopoietin levels in patients with CAMT are always elevated
of function of and signaling from the thrombopoietin receptor in and are among the highest seen in any patient population.
type I by deletion of all or most of the intracellular domain. This The pathogenesis of the associated neurologic abnormalities (see
causes persistently low platelet counts and a rapid progression to Clinical Features) is less understood; however, MPL is expressed in
pancytopenia. Thrombopoietin plays a critical role in the prolifera- the neuronal cells and might be important for their development.
tion, survival, and differentiation of early and late megakaryocytes.
This clearly explains the thrombocytopenia. However, MPL is also
highly expressed in HSCs and promotes their quiescence and survival. Clinical Features
Thus MPL protein insufficiency may account for depletion of HSCs
and pancytopenia. Evolution into severe aplastic anemia is particu- Almost all patients present with a petechial rash, bruising, or bleeding
larly common in type I. during the first year of life. Most cases are obvious at birth or within
the first 2 months. Most patients with proven MPL mutations have
Type II normal physical and imaging features, but isolated cases with anoma-
CAMT with missense and certain splicing mutations cause reduced lies have been identified. Many of the published cases with CAMT
expression of the protein, reduced localization to the plasma mem- and physical malformation were not tested for MPL mutations. A
brane (e.g., R102P in the extracellular domain), or an inability to patient in the CIMFR with an MPL mutation had a cystic fourth
bind thrombopoietin (e.g., F104S). Patients with these mutations ventricle and Dandy-Walker malformation (Dror, unpublished data).
have a milder course; a transient increase in platelet counts during The commonest anomalies in published phenotypic CAMT patients
the first years of life; and delayed onset, if any, of pancytopenia, are neurologic, including varying degrees of cerebellar hypoplasia or
indicating residual receptor function. agenesis, cerebral atrophy, cortical dysplasia and lissencephaly, and
Serial studies of CAMT hematopoiesis using clonogenic assays hypoplasia of the corpus callosum and brainstem. Facial malforma-
have been informative. Initially, when the only hematologic abnor- tions have also been described. Developmental delay is a prominent
mality is isolated thrombocytopenia, the numbers of hematopoietic feature among those with physical malformations. Patients may also
progenitors are comparable to those of control participants, including have microcephaly and an abnormal facies.
the number of megakaryocyte precursors, CFU-MK (colony-forming Congenital heart disease with a variety of malformations can be
unit megakaryocytes). As the disease evolves into aplastic anemia, the detected, including atrial septal defects, ventricular septal defects,
peripheral blood counts decline, and colony numbers from progeni- patent ductus arteriosus, tetralogy of Fallot, and coarctation of the
tors belonging to each myeloid lineage also decline in parallel. Stromal aorta. Some of these occur in combinations. Other anomalies include
cells established in short- and long-term cultures of patient BM show abnormal hips or feet, kidney malformations, eye anomalies, and cleft
normal proliferative activity and yield a “fertile” BM microenviron- or high-arched palate. Some affected sibships manifested both normal
ment for patient and control BM colony growth. The findings are and abnormal physical findings in the same family.
consistent with current knowledge about MPL mutations, namely,
that the central problem in CAMT is an intrinsic HSC defect rather
than an abnormality of the BM milieu. Laboratory Findings
Other data demonstrate measurable numbers of CFU-MK pro-
genitors in vitro from patients with CAMT when studied early in the Thrombocytopenia is the major laboratory finding with normal
disease in response to IL-3, GM-CSF, or a combination of both but hemoglobin levels and white blood cell counts initially. Although
defective CFU-MK colony formation in response to recombinant there are usually measurable but reduced platelet numbers, peripheral