
Page 470 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 470
Chapter 29 Inherited Bone Marrow Failure Syndromes 391
Etiology and Pathophysiology
Thrombocytopenia in TAR syndrome is the result of a defect in
megakaryocytopoiesis and thrombocytopoiesis. It was previously
shown that patients with TAR have submicroscopic deletions at
1q21.1 in one allele of RBMA8 in 100% of cases. The allele carries
one of two low-frequency SNPs in the regulatory regions of RBM8A.
RBM8A encodes the Y14 subunit of exon-junction complex, which
processes mRNA. The mutations caused reduced expression of the
protein in platelets from affected individuals.
Thrombopoietin levels in plasma or serum are consistently elevated
in TAR syndrome, thereby excluding a cytokine production defect as
a cause for thrombocytopenia in this disorder. BM CFU-MK pro-
genitors are either absent or are present in low to normal frequencies
but produce small colonies in vitro with abnormal morphology.
CFU-GM and BFU-E colony growth is often increased.
+
In a detailed study of CD34 cells, the thrombocytopenia of TAR
syndrome was associated with a dysmegakaryocytopoiesis character-
ized by cells remaining at an early stage of differentiation. Cells
expressing CD41 without CD42 accumulated behind the block, and
there was a decrease in c-mpl transcripts and mpl protein. The
response of platelets to adenosine diphosphate or to the thrombin
receptor agonist peptide SFLLRN (TRAP) is normal in patients with
TAR. However, in contrast to control participants, platelets from
patients with TAR do not undergo activation in vitro in response to
recombinant thrombopoietin as measured by testing thrombopoietin
synergism to adenosine diphosphate and TRAP. Thrombopoietin-
induced tyrosine phosphorylation of platelet proteins in this setting
is completely absent or markedly decreased. The results indicate that
there is a lack of response to thrombopoietin downstream the c-mpl
signal transduction pathway.
No recurrent chromosomal changes are seen in TAR syndrome.
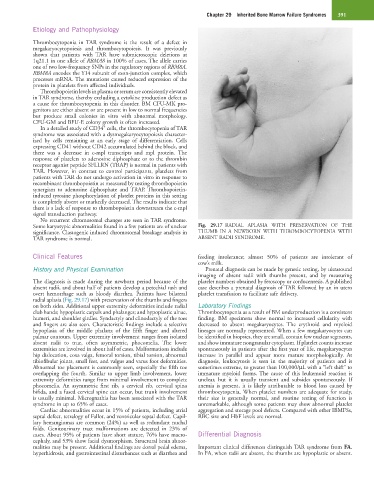
Some karyotypic abnormalities found in a few patients are of unclear Fig. 29.17 RADIAL APLASIA WITH PRESERVATION OF THE
significance. Clastogenic induced chromosomal breakage analysis in THUMB IN A NEWBORN WITH THROMBOCYTOPENIA WITH
TAR syndrome is normal. ABSENT RADII SYNDROME.
Clinical Features feeding intolerance; almost 50% of patients are intolerant of
cow’s milk.
History and Physical Examination Prenatal diagnosis can be made by genetic testing, by ultrasound
imaging of absent radii with thumbs present, and by measuring
The diagnosis is made during the newborn period because of the platelet numbers obtained by fetoscopy or cordocentesis. A published
absent radii, and about half of patients develop a petechial rash and case describes a prenatal diagnosis of TAR followed by an in utero
overt hemorrhage such as bloody diarrhea. Patients have bilateral platelet transfusion to facilitate safe delivery.
radial aplasia (Fig. 29.17) with preservation of the thumbs and fingers
on both sides. Additional upper extremity deformities include radial Laboratory Findings
club hands; hypoplastic carpals and phalanges; and hypoplastic ulnae, Thrombocytopenia as a result of BM underproduction is a consistent
humeri, and shoulder girdles. Syndactyly and clinodactyly of the toes finding. BM specimens show normal to increased cellularity with
and fingers are also seen. Characteristic findings include a selective decreased to absent megakaryocytes. The erythroid and myeloid
hypoplasia of the middle phalanx of the fifth finger and altered lineages are normally represented. When a few megakaryocytes can
palmar contours. Upper extremity involvement ranges from isolated be identified in biopsies, they are small, contain few nuclear segments,
absent radii to true, often asymmetric, phocomelia. The lower and show immature nongranular cytoplasm. If platelet counts increase
extremities are involved in about half of cases. Malformations include spontaneously in patients after the first year of life, megakaryocytes
hip dislocation, coxa valga, femoral torsion, tibial torsion, abnormal increase in parallel and appear more mature morphologically. At
tibiofibular joints, small feet, and valgus and varus foot deformities. diagnosis, leukocytosis is seen in the majority of patients and is
Abnormal toe placement is commonly seen, especially the fifth toe sometimes extreme, to greater than 100,000/µL with a “left shift” to
overlapping the fourth. Similar to upper limb involvement, lower immature myeloid forms. The cause of this leukemoid reaction is
extremity deformities range from minimal involvement to complete unclear, but it is usually transient and subsides spontaneously. If
phocomelia. An asymmetric first rib, a cervical rib, cervical spina anemia is present, it is likely attributable to blood loss caused by
bifida, and a fused cervical spine can occur, but trunk involvement thrombocytopenia. When platelet numbers are adequate for study,
is usually minimal. Micrognathia has been associated with the TAR their size is generally normal, and routine testing of function is
syndrome in up to 65% of cases. unremarkable, although some patients may show abnormal platelet
Cardiac abnormalities occur in 15% of patients, including atrial aggregation and storage pool defects. Compared with other IBMFSs,
septal defect, tetralogy of Fallot, and ventricular septal defect. Capil- RBC size and HbF levels are normal.
lary hemangiomas are common (24%) as well as redundant nuchal
folds. Genitourinary tract malformations are detected in 23% of
cases. About 95% of patients have short stature, 76% have macro- Differential Diagnosis
cephaly, and 53% show facial dysmorphism. Structural brain abnor-
malities may be present. Additional findings are dorsal pedal edema, Important clinical differences distinguish TAR syndrome from FA.
hyperhidrosis, and gastrointestinal disturbances such as diarrhea and In FA, when radii are absent, the thumbs are hypoplastic or absent.