
Page 648 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 648
550 Part V Red Blood Cells
α-Globin α Precipitates Membrane damage
α α α α α α α of α-globin Abnormal metabolism
Inclusion bodies
α-Gene α mRNA α α α α α β + α α α in RBC precursors
2 2
α α α
β-Gene β mRNA β β Hb A Excess α-globin
β-Globin
1 Hb per cell produced
(hypochromia) Massive death of RBC
2 Massive mature RBC precursors in bone marrow
production (inneffective erythropoiesis)
3 Shortened RBC survival
Few surviving RBCs are
highly abnormal,
carry inclusions
Sequestration Bizarre
in spleen morphology
Splenomegaly hypersplenism
Hb catabolism bilirubin
Erythropoietin Tissue hypoxia High-output heart failure,
released by Profound anemia infection, leg ulcers, pallor,
kidney growth retardation
Jaundice
Gallstones
Transfusion Leg ulcers
Bony deformities, fractures,
Massive expansion extramedullary hematopoiesis
of bone marrow Iron overload and Cirrhosis
Paryenchymal
Increased gastrointestinal iron deposition Endocrine dysfunction
iron absorption Cardiomyopathy
(hemochromatosis)
Increased blood volume,
secondary folate deficiency,
pathologic bone fractures
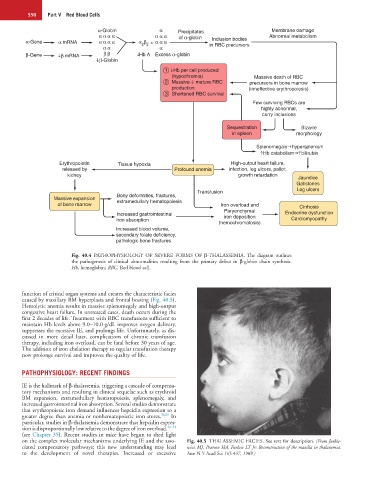
Fig. 40.4 PATHOPHYSIOLOGY OF SEVERE FORMS OF β-THALASSEMIA. The diagram outlines
the pathogenesis of clinical abnormalities resulting from the primary defect in β-globin chain synthesis.
Hb, hemoglobin; RBC, Red blood cell.
function of critical organ systems and creates the characteristic facies
caused by maxillary BM hyperplasia and frontal bossing (Fig. 40.5).
Hemolytic anemia results in massive splenomegaly and high-output
congestive heart failure. In untreated cases, death occurs during the
first 2 decades of life. Treatment with RBC transfusions sufficient to
maintain Hb levels above 9.0–10.0 g/dL improves oxygen delivery,
suppresses the excessive IE, and prolongs life. Unfortunately, as dis-
cussed in more detail later, complications of chronic transfusion
therapy, including iron overload, can be fatal before 30 years of age.
The addition of iron chelation therapy to regular transfusion therapy
now prolongs survival and improves the quality of life.
PATHOPHYSIOLOGY: RECENT FINDINGS
IE is the hallmark of β-thalassemia, triggering a cascade of compensa-
tory mechanisms and resulting in clinical sequelae such as erythroid
BM expansion, extramedullary hematopoiesis, splenomegaly, and
increased gastrointestinal iron absorption. Several studies demonstrate
that erythropoietic iron demand influences hepcidin expression to a
greater degree than anemia or nonhematopoietic iron stores. 38,39 In
particular, studies in β-thalassemia demonstrate that hepcidin expres-
sion is disproportionally low relative to the degree of iron overload. 40–42
(see Chapter 35). Recent studies in mice have begun to shed light
on the complex molecular mechanisms underlying IE and the asso- Fig. 40.5 THALASSEMIC FACIES. See text for description. (From Jurkie-
ciated compensatory pathways; this new understanding may lead wicz MJ, Pearson HA, Furlow LT Jr: Reconstruction of the maxilla in thalassemia.
to the development of novel therapies. Increased or excessive Ann N Y Acad Sci 165:437, 1969.)