
Page 556 - Williams Hematology ( PDFDrive )
P. 556
530 Part VI: The Erythrocyte Chapter 35: Aplastic Anemia: Acquired and Inherited 531
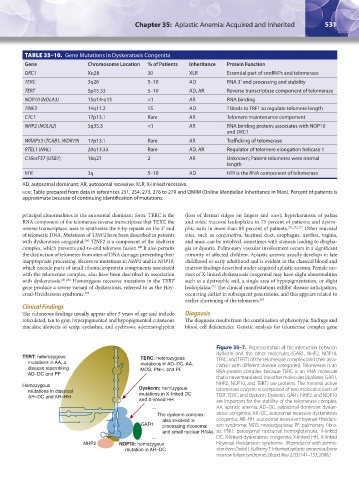
TABLE 35–10. Gene Mutations in Dyskeratosis Congenita
Gene Chromosome Location % of Patients Inheritance Protein Function
DKC1 Xq28 30 XLR Essential part of snoRNPs and telomerase
TERC 3q26 5–10 AD RNA 3′ end processing and stability
TERT 5p15.33 5–10 AD, AR Reverse transcriptase component of telomerase
NOP10 (NOLA3) 15q14-q15 <1 AR RNA binding
TINF2 14q11.2 15 AD ? Binds to TRF1 to regulate telomere length
CTC1 17p13.1 Rare AR Telomere maintenance component
NHP2 (NOLA2) 5q35.3 <1 AR RNA binding protein; associates with NOP10
and DKC1
WRAP53 (TCAB1, WDR79) 17p13.1 Rare AR Trafficking of telomerase
RTEL1 (NHL) 20q13.33 Rare AD, AR Regulator of telomere elongation helicase 1
C16orF57 (USB1) 16q21 2 AR Unknown; Patient telomeres were normal
length
hTR 3q 5–10 AD hTR is the RNA component of telomerase
AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive.
note: Table prepared from data in references 251, 254, 273, 276 to 279 and OMIM (Online Mendelian Inheritance in Man). Percent of patients is
approximate because of continuing identification of mutations.
principal abnormalities in the autosomal dominant form. TERC is the (loss of dermal ridges on fingers and toes); hyperkeratosis of palms
RNA component of the telomerase reverse transcriptase that TERT, the and soles; mucosal leukoplakia in 75 percent of patients; and dystro-
reverse transcriptase, uses to synthesize the 6-bp repeats on the 3′ end phic nails in more than 85 percent of patients. 251,272,273 Other mucosal
of telomeric DNA. Mutations of TINF2 have been described in patients sites, such as conjunctiva, lacrimal duct, esophagus, urethra, vagina,
with dyskeratosis congenital. TINF2 is a component of the shelterin and anus, can be involved, sometimes with stenosis leading to dyspha-
280
complex, which prevents end-to-end telomere fusion. It also permits gia or dysuria. Pulmonary vascular involvement occurs in a significant
280
the distinction of telomeres from sites of DNA damage, preventing their minority of affected children. Aplastic anemia usually develops in late
inappropriate processing. Recessive mutations in NHP2 and in NOP10, childhood or early adulthood and is evident in the classical blood and
which encode parts of small ribonucleoprotein components associated marrow findings described under acquired aplastic anemia. Female car-
with the telomerase complex, also have been described in association riers of X-linked dyskeratosis congenital may have slight abnormalities
with dyskeratosis. 281,282 Homozygous recessive mutations in the TERT such as a dystrophic nail, a single area of hypopigmentation, or slight
272
gene produce a severe variant of dyskeratosis, referred to as the Hoy- leukoplakia. The clinical manifestations exhibit disease anticipation,
eraal-Hreidarsson syndrome. 272 occurring earlier in subsequent generations, and this appears related to
earlier shortening of the telomeres. 283
Clinical Findings
The cutaneous findings usually appear after 5 years of age and include Diagnosis
reticulated, tan to gray, hyperpigmented and hypopigmented cutaneous The diagnosis results from the combination of phenotypic findings and
macules; alopecia of scalp, eyelashes, and eyebrows; adermatoglyphia blood cell deficiencies. Genetic analysis for telomerase complex gene
Figure 35–7. Representation of the interaction between
dyskerin and the other molecules (GAR1, NHP2, NOP10,
TERT: heterozygous TERC: heterozygous TERC, and TERT) of the telomerase complex (and their asso-
mutations in AA, a mutations in AD–DC, AA, ciation with different disease categories). Telomerase is an
disease resembling MDS, PNH, and PF RNA-protein complex because TERC is an RNA molecule
AD–DC and PF
that is never translated. The other molecules (dyskerin, GAR1,
Homozygous Dyskerin: hemizygous NHP2, NOP10, and TERT) are proteins. The minimal active
mutations in classical mutations in X-linked DC telomerase enzyme is composed of two molecules each of
AR–DC and AR–HH TERT, TERC, and dyskerin. Dyskerin, GAR1, NHP2, and NOP10
and X-linked HH are important for the stability of the telomerase complex.
AA, aplastic anemia; AD–DC, autosomal dominant dysker-
The dyskerin complex: atosis congenita; AR–DC, autosomal recessive dyskeratosis
also involved in congenita; AR–HH, autosomal recessive Hoyeraal-Hreidars-
GAR1 processing ribosomal son syndrome; MDS, myelodysplasia; PF, pulmonary fibro-
and small nuclear RNAs sis; PNH, paroxysmal nocturnal hemoglobinuria; X-linked
DC, X-linked dyskeratosis congenita; X-linked HH, X-linked
NHP2 NOP10: homozygous Hoyeraal-Hreidarsson syndrome. (Reproduced with permis-
mutation in AR–DC sion from Dokal I, Vulliamy T: Inherited aplastic anaemias/bone
marrow failure syndromes. Blood Rev 22(3):141–153, 2008.)
Kaushansky_chapter 35_p0513-0538.indd 531 9/19/15 12:24 AM