
Page 522 - Clinical Immunology_ Principles and Practice ( PDFDrive )
P. 522
502 ParT fOur Immunological Deficiencies
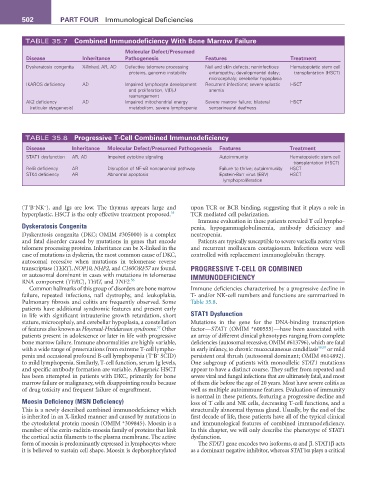
TABLE 35.7 Combined Immunodeficiency With Bone Marrow failure
Molecular Defect/Presumed
Disease Inheritance Pathogenesis features Treatment
Dyskeratosis congenita X-linked, AR, AD Defective telomere processing Nail and skin defects; noninfectious Hematopoietic stem cell
proteins, genomic instability enteropathy; developmental delay; transplantation (HSCT)
microcephaly; cerebellar hypoplasia
IKAROS deficiency AD Impaired lymphocyte development Recurrent infections; severe aplastic HSCT
and proliferation, V(D)J anemia
rearrangement
AK2 deficiency AD Impaired mitochondrial energy Severe marrow failure; bilateral HSCT
(reticular dysgenesis) metabolism, severe lymphopenia sensorineural deafness
TABLE 35.8 Progressive T-Cell Combined Immunodeficiency
Disease Inheritance Molecular Defect/Presumed Pathogenesis features Treatment
STAT1 dysfunction AR, AD Impaired cytokine signaling Autoimmunity Hematopoietic stem cell
transplantation (HSCT)
RelB deficiency AR Disruption of NF-κB noncanonical pathway Failure to thrive; autoimmunity HSCT
STK4 deficiency AR Abnormal apoptosis Epstein-Barr virus (EBV) HSCT
lymphoproliferation
−
+ −
(T B NK ), and Igs are low. The thymus appears large and upon TCR or BCR binding, suggesting that it plays a role in
hyperplastic. HSCT is the only effective treatment proposed. 55 TCR mediated cell polarization.
Immune evaluation in these patients revealed T cell lympho-
Dyskeratosis Congenita penia, hypogammaglobulinemia, antibody deficiency and
Dyskeratosis congenita (DKC; OMIM #305000) is a complex neutropenia.
and fatal disorder caused by mutations in genes that encode Patients are typically susceptible to severe varicella zoster virus
telomere processing proteins. Inheritance can be X-linked in the and recurrent molluscum contagiosum. Infections were well
case of mutations in dyskerin, the most common cause of DKC, controlled with replacement immunoglobulin therapy.
autosomal recessive when mutations in telomerase reverse
transcriptase (TERT), NOP10, NHP2, and C160ORF57 are found, PROGRESSIVE T-CELL OR COMBINED
or autosomal dominant in cases with mutations in telomerase IMMUNODEFICIENCY
RNA component (TERC), TERT, and TNF2. 56
Common hallmarks of this group of disorders are bone marrow Immune deficiencies characterized by a progressive decline in
failure, repeated infections, nail dystrophy, and leukoplakia. T- and/or NK-cell numbers and functions are summarized in
Pulmonary fibrosis and colitis are frequently observed. Some Table 35.8.
patients have additional syndromic features and present early
in life with significant intrauterine growth retardation, short STAT1 Dysfunction
stature, microcephaly, and cerebellar hypoplasia, a constellation Mutations in the gene for the DNA-binding transcription
57
of features also known as Hoyeraal-Hreidarsson syndrome. Other factor—STAT1 (OMIM *600555)—have been associated with
patients present in adolescence or later in life with progressive an array of different clinical phenotypes ranging from complete
bone marrow failure. Immune abnormalities are highly variable, deficiencies (autosomal recessive; OMIM #613796), which are fatal
with a wide range of presentations from extreme T-cell lympho- in early infancy, to chronic mucocutaneous candidiasis 58,59 or mild
− −
penia and occasional profound B-cell lymphopenia (T B SCID) persistent oral thrush (autosomal dominant; OMIM #614892).
to mild lymphopenia. Similarly, T-cell function, serum Ig levels, One subgroup of patients with monoallelic STAT1 mutations
and specific antibody formation are variable. Allogeneic HSCT appear to have a distinct course. They suffer from repeated and
has been attempted in patients with DKC, primarily for bone severe viral and fungal infections that are ultimately fatal, and most
marrow failure or malignancy, with disappointing results because of them die before the age of 20 years. Most have severe colitis as
of drug toxicity and frequent failure of engraftment. well as multiple autoimmune features. Evaluation of immunity
is normal in these patients, featuring a progressive decline and
Moesin Deficiency (MSN Deficiency) loss of T cells and NK cells, decreasing T-cell functions, and a
This is a newly described combined immunodeficiency which structurally abnormal thymus gland. Usually, by the end of the
is inherited in an X-linked manner and caused by mutations in first decade of life, these patients have all of the typical clinical
the cytoskeletal protein moesin (OMIM *309845). Moesin is a and immunological features of combined immunodeficiency.
member of the ezrin-radixin-moesin family of proteins that link In this chapter, we will only describe the phenotype of STAT1
the cortical actin filaments to the plasma membrane. The active dysfunction.
form of moesin is predominantly expressed in lymphocytes where The STAT1 gene encodes two isoforms, α and β. STAT1β acts
it is believed to sustain cell shape. Moesin is dephosphorylated as a dominant negative inhibitor, whereas STAT1α plays a critical