
Page 2117 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 2117
1880 Part XII Hemostasis and Thrombosis
Platelet Aggregation Testing for the Diagnosis of Platelet Disorders
Platelet aggregometry is the mainstay in the work-up of platelet func-
tion disorders (PFDs). Abnormalities in aggregation tracings can point
towards specific diagnoses and direct further testing.
Light Transmission Glanzmann Thrombasthenia
In this severe PFD in which there are quantitative or qualitative defects
in integrin αIIbβ3 (GPIIb–IIIa), the receptor to which fibrinogen/von
Willebrand factor (VWF) binds on activated platelets, there is absent
aggregation response to all agonists. However, agglutination in response
to a high concentration of ristocetin occurs, as it is independent of
αIIbβ3.
Bernard-Soulier Syndrome
This disorder with defective surface expression of the adhesive VWF
receptor GPIb–IX–V is characterized by a lack of agglutination to a high
concentration of ristocetin. Note that in von Willebrand disease (VWD)
2.3 µM 4.5 µM 9.1 µM there is an absent agglutination response to a high concentration of
ristocetin because of deficient VWF.
Time Defects in Agonist/Adhesion Receptors
1 min
P2Y12: a defect in this ADP receptor results in decreased and reversible
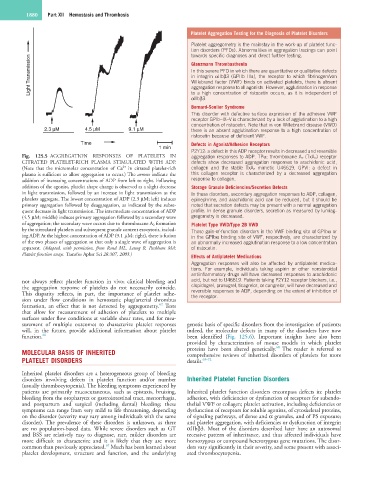
Fig. 125.5 AGGREGATION RESPONSES OF PLATELETS IN aggregation responses to ADP. TPα: thromboxane A 2 (TxA 2 ) receptor
CITRATED PLATELET-RICH PLASMA STIMULATED WITH ADP. defects show decreased aggregation responses to arachidonic acid,
2+
(Note that the micromolar concentration of Ca in citrated platelet-rich collagen and the stable TxA 2 mimetic U46619. GPVI: a defect in
plasma is sufficient to allow aggregation to occur.) The arrows indicate the this collagen receptor is characterized by a decreased aggregation
addition of increasing concentrations of ADP from left to right. Following response to collagen.
addition of the agonist, platelet shape change is observed as a slight decrease Storage Granule Deficiencies/Secretion Defects
in light transmission, followed by an increase in light transmission as the In these disorders, secondary aggregation responses to ADP, collagen,
platelets aggregate. The lowest concentration of ADP (2.3 µM; left) induces epinephrine, and arachidonic acid can be reduced, but it should be
primary aggregation followed by deaggregation, as indicated by the subse- noted that secretion defects may be present with a normal aggregation
quent decrease in light transmission. The intermediate concentration of ADP profile. In dense granule disorders, secretion as measured by lumiag-
(4.5 µM; middle) induces primary aggregation followed by a secondary wave gregometry is decreased.
of aggregation; the secondary wave occurs due to thromboxane A 2 formation Platelet Type VWD/Type 2B VWD
by the stimulated platelets and subsequent granule content exocytosis, includ- These gain-of-function disorders in the VWF binding site of GPIbα or
ing ADP. At the highest concentration of ADP (9.1 µM; right), there is fusion in the GPIbα binding site of VWF, respectively, are characterized by
of the two phases of aggregation so that only a single wave of aggregation is an abnormally increased agglutination response to a low concentration
apparent. (Adapted, with permission, from Rand ML, Leung R, Packham MA: of ristocetin.
Platelet function assays. Transfus Apher Sci 28:307, 2003.)
Effects of Antiplatelet Medications
Aggregation responses will also be affected by antiplatelet medica-
tions. For example, individuals taking aspirin or other nonsteroidal
antiinflammatory drugs will have decreased responses to arachidonic
not always reflect platelet function in vivo; clinical bleeding and acid, but not to U46619. Patients taking P2Y12 receptor blockers, i.e.,
the aggregation response of platelets do not necessarily coincide. clopidogrel, prasugrel, ticagrelor, or cangrelor, will have decreased and
This disparity reflects, in part, the importance of platelet adhe- reversible responses to ADP, depending on the extent of inhibition of
the receptor.
sion under flow conditions in hemostatic plug/arterial thrombus
62
formation, an effect that is not detected by aggregometry. Tests
that allow for measurement of adhesion of platelets to multiple
surfaces under flow conditions at variable shear rates, and for mea-
surement of multiple outcomes to characterize platelet responses genetic basis of specific disorders from the investigation of patients;
will, in the future, provide additional information about platelet indeed, the molecular defects in many of the disorders have now
function. 66 been identified (Fig. 125.6). Important insights have also been
provided by characterization of mouse models in which platelet
68
MOLECULAR BASIS OF INHERITED proteins have been altered genetically. The reader is referred to
comprehensive reviews of inherited disorders of platelets for more
PLATELET DISORDERS details. 69–72
Inherited platelet disorders are a heterogeneous group of bleeding
disorders involving defects in platelet function and/or number Inherited Platelet Function Disorders
(usually thrombocytopenia). The bleeding symptoms experienced by
patients are primarily mucocutaneous, such as epistaxis, bruising, Inherited platelet function disorders encompass defects in: platelet
bleeding from the oropharynx or gastrointestinal tract, menorrhagia, adhesion, with deficiencies or dysfunction of receptors for subendo-
and postpartum and surgical (including dental) bleeding; these thelial VWF or collagen; platelet activation, including deficiencies or
symptoms can range from very mild to life threatening, depending dysfunction of receptors for soluble agonists, of cytoskeletal proteins,
on the disorder (severity may vary among individuals with the same of signaling pathways, of dense and α granules, and of PS exposure;
disorder). The prevalence of these disorders is unknown, as there and platelet aggregation, with deficiencies or dysfunction of integrin
are no population-based data. While severe disorders such as GT αIIbβ3. Most of the disorders described later have an autosomal
and BSS are relatively easy to diagnose, rare, milder disorders are recessive pattern of inheritance, and thus affected individuals have
more difficult to characterize and it is likely that they are more homozygous or compound heterozygous gene mutations. The disor-
67
common than previously appreciated. Much has been learned about ders vary significantly in their severity, and some present with associ-
platelet development, structure and function, and the underlying ated thrombocytopenia.