
Page 977 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 977
860 Part VII Hematologic Malignancies
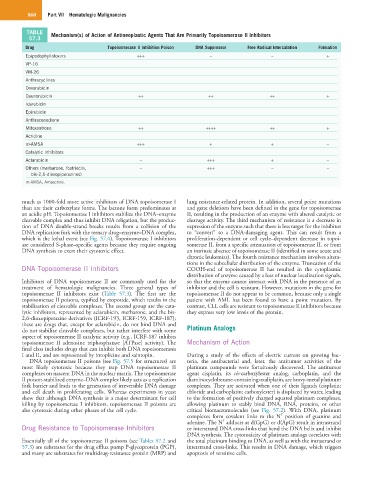
TABLE Mechanism(s) of Action of Antineoplastic Agents That Are Primarily Topoisomerase II Inhibitors
57.3
Drug Topoisomerase II Inhibition Poison DNA Suppressor Free Radical Intercalation Formation
Epipodophyllotoxins +++ – – +
VP-16
VM-26
Anthracyclines
Doxorubicin
Daunorubicin ++ ++ ++ +
Idarubicin
Epirubicin
Anthracenedione
Mitoxantrone ++ ++++ ++ +
Acridine
m-AMSA +++ + + –
Catalytic inhibitors
Aclarubicin – +++ + –
Others (merbarone, fostriecin, – +++ – –
bis-2,6-dioxopiperazines)
m-AMSA, Amsacrine.
much as 1000-fold more active inhibitors of DNA topoisomerase I lung resistance-related protein. In addition, several point mutations
than are their carboxylate forms. The lactone form predominates at and gene deletions have been defined in the gene for topoisomerase
an acidic pH. Topoisomerase I inhibitors stabilize the DNA–enzyme II, resulting in the production of an enzyme with altered catalytic or
cleavable complex and thus inhibit DNA religation, but the produc- cleavage activity. The third mechanism of resistance is a decrease in
tion of DNA double-strand breaks results from a collision of the expression of the enzyme such that there is less target for the inhibitor
DNA replication fork with the ternary drug–enzyme–DNA complex, to “convert” to a DNA-damaging agent. This can result from a
which is the lethal event (see Fig. 57.4). Topoisomerase I inhibitors proliferation-dependent or cell cycle–dependent decrease in topoi-
are considered S-phase–specific agents because they require ongoing somerase II, from a specific attenuation of topoisomerase II, or from
DNA synthesis to exert their cytotoxic effect. an intrinsic absence of topoisomerase II (identified in some acute and
chronic leukemias). The fourth resistance mechanism involves altera-
tions in the subcellular distribution of the enzyme. Truncation of the
DNA Topoisomerase II Inhibitors COOH-end of topoisomerase II has resulted in the cytoplasmic
distribution of enzyme caused by a loss of nuclear localization signals,
Inhibitors of DNA topoisomerase II are commonly used for the so that the enzyme cannot interact with DNA in the presence of an
treatment of hematologic malignancies. Three general types of inhibitor and the cell is resistant. However, mutations in the gene for
topoisomerase II inhibitors exist (Table 57.3). The first are the topoisomerase II do not appear to be common, because only a single
topoisomerase II poisons, typified by etoposide, which results in the patient with AML has been found to have a point mutation. By
stabilization of cleavable complexes. The second group are the cata- contrast, CLL cells are resistant to topoisomerase II inhibitors because
lytic inhibitors, represented by aclarubicin, merbarone, and the bis- they express very low levels of the protein.
2,6-dioxopiperazine derivatives (ICRF-193, ICRF-159, ICRF-187);
these are drugs that, except for aclarubicin, do not bind DNA and
do not stabilize cleavable complexes, but rather interfere with some Platinum Analogs
aspect of topoisomerase II catalytic activity (e.g., ICRF-187 inhibits
topoisomerase II adenosine triphosphatase [ATPase] activity). The Mechanism of Action
final class includes drugs that can inhibit both DNA topoisomerases
I and II, and are represented by intoplicine and saintopin. During a study of the effects of electric current on growing bac-
DNA topoisomerase II poisons (see Fig. 57.5 for structures) are teria, the antibacterial and, later, the antitumor activities of the
most likely cytotoxic because they trap DNA topoisomerase II platinum compounds were fortuitously discovered. The antitumor
complexes on nascent DNA in the nuclear matrix. The topoisomerase agent cisplatin, its cis-carboxylester analog, carboplatin, and the
II poison-stabilized enzyme–DNA complex likely acts as a replication diaminocyclohexane-containing oxaliplatin, are heavy-metal platinum
fork barrier and leads to the generation of irreversible DNA damage complexes. They are activated when one of their ligands (cisplatin;
and cell death in proliferating cells. Whereas experiments in yeast chloride and carboplatin; carboxylester) is displaced by water, leading
show that although DNA synthesis is a major determinant for cell to the formation of positively charged aquated platinum complexes,
killing by topoisomerase I inhibitors, topoisomerase II poisons are allowing platinum to stably bind DNA, RNA, proteins, or other
also cytotoxic during other phases of the cell cycle. critical biomacromolecules (see Fig. 57.2). With DNA, platinum
7
complexes form covalent links to the N position of guanine and
7
adenine. The N adducts at d(GpG) or d(ApG) result in intrastrand
Drug Resistance to Topoisomerase Inhibitors or interstrand DNA cross-links that bend the DNA helix and inhibit
DNA synthesis. The cytotoxicity of platinum analogs correlates with
Essentially all of the topoisomerase II poisons (see Tables 57.2 and the total platinum binding to DNA, as well as with the intrastrand or
57.3) are substrates for the drug efflux pump P-glycoprotein (PGP), interstrand cross-links. This results in DNA damage, which triggers
and many are substrates for multidrug-resistance protein (MRP) and apoptosis of sensitive cells.