
Page 1045 - Williams Hematology ( PDFDrive )
P. 1045
1020 Part VII: Neutrophils, Eosinophils, Basophils, and Mast Cells Chapter 66: Disorders of Neutrophil Function 1021
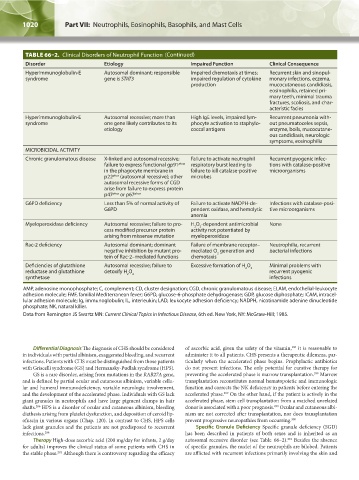
TABLE 66–2. Clinical Disorders of Neutrophil Function (Continued)
Disorder Etiology Impaired Function Clinical Consequence
Hyperimmunoglobulin-E Autosomal dominant; responsible Impaired chemotaxis at times; Recurrent skin and sinopul-
syndrome gene is STAT3 impaired regulation of cytokine monary infections, eczema,
production mucocutaneous candidiasis,
eosinophilia, retained pri-
mary teeth, minimal trauma
fractures, scoliosis, and char-
acteristic facies
Hyperimmunoglobulin-E Autosomal recessive; more than High IgE levels, impaired lym- Recurrent pneumonia with-
syndrome one gene likely contributes to its phocyte activation to staphylo- out pneumatoceles sepsis,
etiology coccal antigens enzyme, boils, mucocutane-
ous candidiasis, neurologic
symptoms, eosinophilia
MICROBICIDAL ACTIVITY
Chronic granulomatous disease X-linked and autosomal recessive; Failure to activate neutrophil Recurrent pyogenic infec-
failure to express functional gp91 phox respiratory burst leading to tions with catalase-positive
in the phagocyte membrane in failure to kill catalase-positive microorganisms
p22 phox (autosomal recessive); other microbes
autosomal recessive forms of CGD
arise from failure to express protein
p47 phox or p67 phox
G6PD deficiency Less than 5% of normal activity of Failure to activate NADPH-de- Infections with catalase-posi-
G6PD pendent oxidase, and hemolytic tive microorganisms
anemia
Myeloperoxidase deficiency Autosomal recessive; failure to pro- H O -dependent antimicrobial None
2
2
cess modified precursor protein activity not potentiated by
arising from missense mutation myeloperoxidase
Rac-2 deficiency Autosomal dominant; dominant Failure of membrane receptor– Neutrophilia, recurrent
negative inhibition by mutant pro- mediated O generation and bacterial infections
2
tein of Rac-2–mediated functions chemotaxis
Deficiencies of glutathione Autosomal recessive; failure to Excessive formation of H O 2 Minimal problems with
2
reductase and glutathione detoxify H O 2 recurrent pyogenic
2
synthetase infections
AMP, adenosine monophosphate; C, complement; CD, cluster designation; CGD, chronic granulomatous disease; ELAM, endothelial-leukocyte
adhesion molecule; FMF, familial Mediterranean fever; G6PD, glucose-6-phosphate dehydrogenase; GDP, glucose diphosphate; ICAM, intracel-
lular adhesion molecule; Ig, immunoglobulin; IL, interleukin; LAD, leukocyte adhesion deficiency; NADPH, nicotinamide adenine dinucleotide
phosphate; NK, natural killer.
Data from Remington JS Swartz MN: Current Clinical Topics in Infectious Disease, 6th ed. New York, NY: McGraw-Hill; 1985.
286
Differential Diagnosis The diagnosis of CHS should be considered of ascorbic acid, given the safety of the vitamin, it is reasonable to
in individuals with partial albinism, exaggerated bleeding, and recurrent administer it to all patients. CHS presents a therapeutic dilemma, par-
infections. Patients with CHS must be distinguished from those patients ticularly when the accelerated phase begins. Prophylactic antibiotics
with Griscelli syndrome (GS) and Hermansky-Pudlak syndrome (HPS). do not prevent infections. The only potential for curative therapy for
GS is a rare disorder, arising from mutations in the RAB27A gene, preventing the accelerated phase is marrow transplantation. Marrow
299
and is defined by partial ocular and cutaneous albinism, variable cellu- transplantation reconstitutes normal hematopoietic and immunologic
lar and humeral immunodeficiency, variable neurologic involvement, function and corrects the NK deficiency in patients before entering the
299
and the development of the accelerated phase. Individuals with GS lack accelerated phase. On the other hand, if the patient is actively in the
giant granules in neutrophils and have large pigment clumps in hair accelerated phase, stem cell transplantation from a matched unrelated
299
286
shafts. HPS is a disorder of ocular and cutaneous albinism, bleeding donor is associated with a poor prognosis. Ocular and cutaneous albi-
diathesis arising from platelet dysfunction, and deposition of ceroid lip- nism are not corrected after transplantation, nor does transplantation
ofuscin in various organs (Chap. 120). In contrast to CHS, HPS cells prevent progressive neuropathies from occurring. 300
lack giant granules and the patients are not predisposed to recurrent Specific Granule Deficiency Specific granule deficiency (SGD)
infections. 286 has been described in patients of both sexes and is inherited as an
281
Therapy High-dose ascorbic acid (200 mg/day for infants, 2 g/day autosomal recessive disorder (see Table 66–2). Besides the absence
for adults) improves the clinical status of some patients with CHS in of specific granules, the nuclei of the neutrophils are bilobed. Patients
281
the stable phase. Although there is controversy regarding the efficacy are afflicted with recurrent infections primarily involving the skin and
Kaushansky_chapter 66_p1005-1042.indd 1020 9/21/15 10:48 AM