
Page 2011 - Williams Hematology ( PDFDrive )
P. 2011
1986 Part XII: Hemostasis and Thrombosis Chapter 116: Classification, Clinical Manifestations, and Evaluation of Disorders of Hemostasis 1987
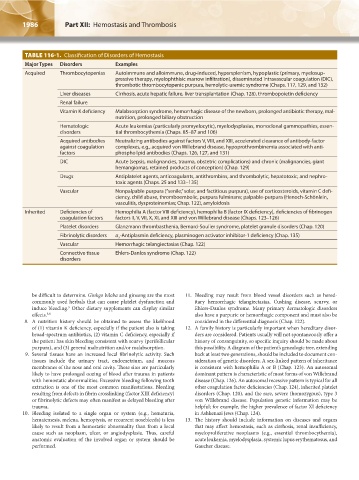
TABLE 116-1. Classification of Disorders of Hemostasis
Major Types Disorders Examples
Acquired Thrombocytopenias Autoimmune and alloimmune, drug-induced, hypersplenism, hypoplastic (primary, myelosup-
pressive therapy, myelophthisic marrow infiltration), disseminated intravascular coagulation (DIC),
thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome (Chaps. 117, 129, and 132)
Liver diseases Cirrhosis, acute hepatic failure, liver transplantation (Chap. 128), thrombopoietin deficiency
Renal failure
Vitamin K deficiency Malabsorption syndrome, hemorrhagic disease of the newborn, prolonged antibiotic therapy, mal-
nutrition, prolonged biliary obstruction
Hematologic Acute leukemias (particularly promyelocytic), myelodysplasias, monoclonal gammopathies, essen-
disorders tial thrombocythemia (Chaps. 85–87 and 106)
Acquired antibodies Neutralizing antibodies against factors V, VIII, and XIII, accelerated clearance of antibody-factor
against coagulation complexes, e.g., acquired von Willebrand disease, hypoprothrombinemia associated with anti-
factors phospholipid antibodies (Chaps. 126, 127, and 131)
DIC Acute (sepsis, malignancies, trauma, obstetric complications) and chronic (malignancies, giant
hemangiomas, retained products of conception) (Chap. 129)
Drugs Antiplatelet agents, anticoagulants, antithrombins, and thrombolytic, hepatotoxic, and nephro-
toxic agents (Chaps. 25 and 133–135)
Vascular Nonpalpable purpura (“senile,” solar, and factitious purpura), use of corticosteroids, vitamin C defi-
ciency, child abuse, thromboembolic, purpura fulminans; palpable-purpura (Henoch-Schönlein,
vasculitis, dysproteinemias; Chap. 122), amyloidosis
Inherited Deficiencies of Hemophilia A (factor VIII deficiency), hemophilia B (factor IX deficiency), deficiencies of fibrinogen
coagulation factors factors II, V, VII, X, XI, and XIII and von Willebrand disease (Chaps. 123–126)
Platelet disorders Glanzmann thrombasthenia, Bernard-Soulier syndrome, platelet granule disorders (Chap. 120)
Fibrinolytic disorders α -Antiplasmin deficiency, plasminogen activator inhibitor-1 deficiency (Chap. 135)
2
Vascular Hemorrhagic telangiectasias (Chap. 122)
Connective tissue Ehlers-Danlos syndrome (Chap. 122)
disorders
be difficult to determine. Ginkgo biloba and ginseng are the most 11. Bleeding may result from blood vessel disorders such as hered-
commonly used herbals that can cause platelet dysfunction and itary hemorrhagic telangiectasias, Cushing disease, scurvy, or
induce bleeding. Other dietary supplements can display similar Ehlers-Danlos syndrome. Many primary dermatologic disorders
5
effects. 5,6 also have a purpuric or hemorrhagic component and must also be
8. A nutrition history should be obtained to assess the likelihood considered in the differential diagnosis (Chap. 122).
of (1) vitamin K deficiency, especially if the patient also is taking 12. A family history is particularly important when hereditary disor-
broad-spectrum antibiotics, (2) vitamin C deficiency, especially if ders are considered. Patients usually will not spontaneously offer a
the patient has skin bleeding consistent with scurvy (perifollicular history of consanguinity, so specific inquiry should be made about
purpura), and (3) general malnutrition and/or malabsorption. this possibility. A diagram of the patient’s genealogic tree, extending
9. Several tissues have an increased local fibrinolytic activity. Such back at least two generations, should be included to document con-
tissues include the urinary tract, endometrium, and mucous sideration of genetic disorders. A sex-linked pattern of inheritance
membranes of the nose and oral cavity. These sites are particularly is consistent with hemophilia A or B (Chap. 123). An autosomal
likely to have prolonged oozing of blood after trauma in patients dominant pattern is characteristic of most forms of von Willebrand
with hemostatic abnormalities. Excessive bleeding following tooth disease (Chap. 126). An autosomal recessive pattern is typical for all
extraction is one of the most common manifestations. Bleeding other coagulation factor deficiencies (Chap. 124), inherited platelet
resulting from defects in fibrin crosslinking (factor XIII deficiency) disorders (Chap. 120), and the rare, severe (homozygous), type 3
or fibrinolytic defects may often manifest as delayed bleeding after von Willebrand disease. Population genetic information may be
trauma. helpful; for example, the higher prevalence of factor XI deficiency
10. Bleeding isolated to a single organ or system (e.g., hematuria, in Ashkenazi Jews (Chap. 124).
hematemesis, melena, hemoptysis, or recurrent nosebleeds) is less 13. The history should include information on diseases and organs
likely to result from a hemostatic abnormality than from a local that may affect hemostasis, such as cirrhosis, renal insufficiency,
cause such as neoplasm, ulcer, or angiodysplasia. Thus, careful myeloproliferative neoplasms (e.g., essential thrombocythemia),
anatomic evaluation of the involved organ or system should be acute leukemia, myelodysplasia, systemic lupus erythematosus, and
performed. Gaucher disease.
Kaushansky_chapter 116_p1985-1992.indd 1986 9/18/15 10:12 AM