
Page 460 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 460
Chapter 29 Inherited Bone Marrow Failure Syndromes 381
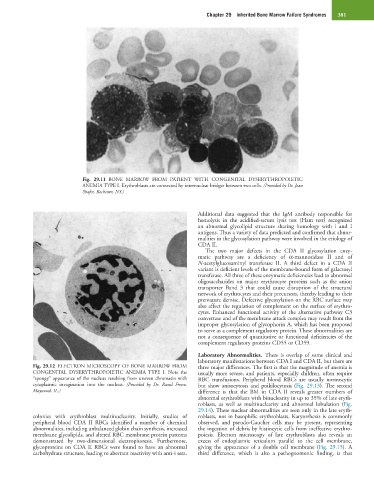
Fig. 29.11 BONE MARROW FROM PATIENT WITH CONGENITAL DYSERYTHROPOIETIC
ANEMIA TYPE I. Erythroblasts are connected by internuclear bridges between two cells. (Provided by Dr. Jean
Shafer, Rochester, NY.)
Additional data suggested that the IgM antibody responsible for
hemolysis in the acidified-serum lysis test (Ham test) recognized
an abnormal glycolipid structure sharing homology with i and I
antigens. Thus a variety of data predicted and confirmed that abnor-
malities in the glycosylation pathway were involved in the etiology of
CDA II.
The two major defects in the CDA II glycosylation enzy-
matic pathway are a deficiency of α-mannosidase II and of
N-acetylglucosaminyl transferase II. A third defect in a CDA II
variant is deficient levels of the membrane-bound form of galactosyl
transferase. All three of these enzymatic deficiencies lead to abnormal
oligosaccharides on major erythrocyte proteins such as the anion
transporter Band 3 that could cause disruption of the structural
network of erythrocytes and their precursors, thereby leading to their
premature demise. Defective glycosylation on the RBC surface may
also affect the regulation of complement on the surface of erythro-
cytes. Enhanced functional activity of the alternative pathway C3
convertase and of the membrane attack complex may result from the
improper glycosylation of glycophorin A, which has been proposed
to serve as a complement regulatory protein. These abnormalities are
not a consequence of quantitative or functional deficiencies of the
complement regulatory proteins CD55 or CD59.
Laboratory Abnormalities. There is overlap of some clinical and
laboratory manifestations between CDA I and CDA II, but there are
Fig. 29.12 ELECTRON MICROSCOPY OF BONE MARROW FROM three major differences. The first is that the magnitude of anemia is
CONGENITAL DYSERYTHROPOIETIC ANEMIA TYPE I. Note the usually more severe, and patients, especially children, often require
“spongy” appearance of the nucleus resulting from uneven chromatin with RBC transfusions. Peripheral blood RBCs are usually normocytic
cytoplasmic invagination into the nucleus. (Provided by Dr. Raoul Fresco, but show anisocytosis and poikilocytosis (Fig. 29.13). The second
Maywood, IL.) difference is that the BM in CDA II reveals greater numbers of
abnormal erythroblasts with binuclearity in up to 35% of late eryth-
roblasts, as well as multinuclearity and abnormal lobulation (Fig.
29.14). These nuclear abnormalities are seen only in the late eryth-
colonies with erythroblast multinuclearity. Initially, studies of roblasts, not in basophilic erythroblasts. Karyorrhexis is commonly
peripheral blood CDA II RBCs identified a number of chemical observed, and pseudo-Gaucher cells may be present, representing
abnormalities, including unbalanced globin chain synthesis, increased the ingestion of debris by histiocytic cells from ineffective erythro-
membrane glycolipids, and altered RBC membrane protein patterns poiesis. Electron microscopy of late erythroblasts also reveals an
demonstrated by two-dimensional electrophoresis. Furthermore, excess of endoplasmic reticulum parallel to the cell membrane,
glycoproteins on CDA II RBCs were found to have an abnormal giving the appearance of a double cell membrane (Fig. 29.15). A
carbohydrate structure, leading to aberrant reactivity with anti-i sera. third difference, which is also a pathognomonic finding, is that