
Page 1538 - Williams Hematology ( PDFDrive )
P. 1538
1512 Part XI: Malignant Lymphoid Diseases Chapter 91: Acute Lymphoblastic Leukemia 1513
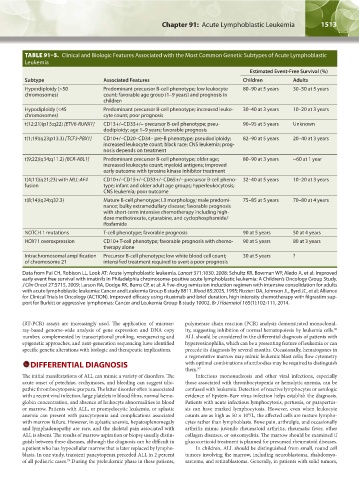
TABLE 91–5. Clinical and Biologic Features Associated with the Most Common Genetic Subtypes of Acute Lymphoblastic
Leukemia
Estimated Event-Free Survival (%)
Subtype Associated Features Children Adults
Hyperdiploidy (>50 Predominant precursor B-cell phenotype; low leukocyte 80–90 at 5 years 30–50 at 5 years
chromosomes) count; favorable age group (1–9 years) and prognosis in
children
Hypodiploidy (<45 Predominant precursor B-cell phenotype; increased leuko- 30–40 at 3 years 10–20 at 3 years
chromosomes) cyte count; poor prognosis
t(12;21)(p13;q22) [ETV6-RUNX1] CD13+/–CD33+/– precursor B-cell phenotype; pseu- 90–95 at 5 years Unknown
dodiploidy; age 1–9 years; favorable prognosis
t(1;19)(q23;p13.3) [TCF3-PBX1] CD10+/–CD20–CD34– pre-B phenotype; pseudodiploidy; 82–90 at 5 years 20–40 at 3 years
increased leukocyte count; black race; CNS leukemia; prog-
nosis depends on treatment
t(9;22)(q34;q11.2) [BCR-ABL1] Predominant precursor B-cell phenotype; older age; 80–90 at 3 years ~60 at 1 year
increased leukocyte count; myeloid antigens; improved
early outcome with tyrosine kinase inhibitor treatment
t(4;11)(q21;23) with MLL-AF4 CD10+/–CD15+/–CD33+/–CD65+/– precursor B-cell pheno- 32–40 at 5 years 10–20 at 3 years
fusion type; infant and older adult age groups; hyperleukocytosis;
CNS leukemia; poor outcome
t(8;14)(q24;q32.3) Mature B-cell phenotype; L3 morphology; male predomi- 75–85 at 5 years 70–80 at 4 years
nance; bulky extramedullary disease; favorable prognosis
with short-term intensive chemotherapy including high-
dose methotrexate, cytarabine, and cyclophosphamide/
ifosfamide
NOTCH 1 mutations T-cell phenotype; favorable prognosis 90 at 5 years 50 at 4 years
HOX11 overexpression CD10+ T-cell phenotype; favorable prognosis with chemo- 90 at 5 years 80 at 3 years
therapy alone
Intrachromosomal amplification Precursor B-cell phenotype; low white blood cell count; 30 at 5 years ?
of chromosome 21 intensified treatment required to avert a poor prognosis
Data from Pui CH, Robison LL, Look AT: Acute lymphoblastic leukemia. Lancet 371:1030, 2008; Schultz KR, Bowman WP, Aledo A, et al: Improved
early event free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: A Children’s Oncology Group Study.
J Clin Oncol 27:5715, 2009; Larson RA, Dodge RK, Burns CP, et al: A five-drug remission induction regimen with intensive consolidation for adults
with acute lymphoblastic leukemia: Cancer and Leukemia Group B study 8811. Blood 85:2025, 1995; Rizzieri DA, Johnson JL, Byrd JC, et al; Alliance
for Clinical Trials In Oncology (ACTION). Improved efficacy using rituximab and brief duration, high intensity chemotherapy with filgrastim sup-
port for Burkitt or aggressive lymphomas: Cancer and Leukemia Group B study 10002. Br J Haematol 165(1):102-111, 2014.
(RT-PCR) assays are increasingly used. The application of microar- polymerase chain reaction (PCR) analysis demonstrated monoclonal-
ray-based genome-wide analysis of gene expression and DNA copy ity, suggesting inhibition of normal hematopoiesis by leukemia cells.
96
number, complemented by transcriptional profiling, resequencing and ALL should be considered in the differential diagnosis of patients with
epigenetic approaches, and next-generation sequencing have identified hypereosinophilia, which can be a presenting feature of leukemia or can
specific genetic alterations with biologic and therapeutic implications. precede its diagnosis by several months. Occasionally, hematogones in
a regenerative marrow may mimic leukemic blast cells; flow cytometry
DIFFERENTIAL DIAGNOSIS with optimal combinations of antibodies may be required to distinguish
them. 97
The initial manifestations of ALL can mimic a variety of disorders. The Infectious mononucleosis and other viral infections, especially
acute onset of petechiae, ecchymoses, and bleeding can suggest idio- those associated with thrombocytopenia or hemolytic anemia, can be
pathic thrombocytopenic purpura. The latter disorder often is associated confused with leukemia. Detection of reactive lymphocytes or serologic
with a recent viral infection, large platelets in blood films, normal hemo- evidence of Epstein-Barr virus infection helps establish the diagnosis.
globin concentration, and absence of leukocyte abnormalities in blood Patients with acute infectious lymphocytosis, pertussis, or parapertus-
or marrow. Patients with ALL, or promyelocytic leukemia, or aplastic sis can have marked lymphocytosis. However, even when leukocyte
anemia can present with pancytopenia and complications associated counts are as high as 50 × 10 /L, the affected cells are mature lympho-
9
with marrow failure. However, in aplastic anemia, hepatosplenomegaly cytes rather than lymphoblasts. Bone pain, arthralgia, and occasionally
and lymphadenopathy are rare, and the skeletal pain associated with arthritis mimic juvenile rheumatoid arthritis, rheumatic fever, other
ALL is absent. The results of marrow aspiration or biopsy usually distin- collagen diseases, or osteomyelitis. The marrow should be examined if
guish between these diseases, although the diagnosis can be difficult in glucocorticoid treatment is planned for presumed rheumatoid diseases.
a patient who has hypocellular marrow that is later replaced by lympho- In children, ALL should be distinguished from small, round cell
blasts. In one study, transient pancytopenia preceded ALL in 2 percent tumors involving the marrow, including neuroblastoma, rhabdomyo-
of all pediatric cases. During the preleukemic phase in these patients, sarcoma, and retinoblastoma. Generally, in patients with solid tumors,
75
Kaushansky_chapter 91_p1505-1526.indd 1513 9/21/15 12:20 PM