
Page 1591 - Williams Hematology ( PDFDrive )
P. 1591
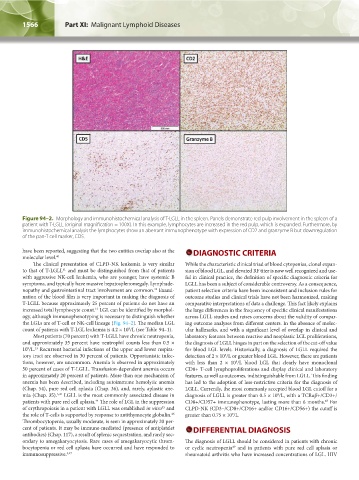
1566 Part XI: Malignant Lymphoid Diseases Chapter 94: Large Granular Lymphocytic Leukemia 1567
Figure 94–2. Morphology and immunohistochemical analysis of T-LGLL in the spleen. Panels demonstrate red pulp involvement in the spleen of a
patient with T-LGLL (original magnification = 100X). In this example, lymphocytes are increased in the red pulp, which is expanded. Furthermore, by
immunohistochemical analysis the lymphocytes show an aberrant immunophenotype with expression of CD2 and granzyme B but downregulation
of the pan-T cell marker, CD5.
have been reported, suggesting that the two entities overlap also at the DIAGNOSTIC CRITERIA
molecular level. 40
The clinical presentation of CLPD-NK leukemia is very similar While the characteristic clinical triad of blood cytopenias, clonal expan-
to that of T-LGLL and must be distinguished from that of patients sion of blood LGL, and elevated RF titer is now well recognized and use-
41
with aggressive NK-cell leukemia, who are younger, have systemic B ful in clinical practice, the definition of specific diagnostic criteria for
symptoms, and typically have massive hepatosplenomegaly. Lymphade- LGLL has been a subject of considerable controversy. As a consequence,
nopathy and gastrointestinal tract involvement are common. Exami- patient selection criteria have been inconsistent and inclusion rules for
42
nation of the blood film is very important in making the diagnosis of outcome studies and clinical trials have not been harmonized, making
T-LGLL because approximately 25 percent of patients do not have an comparative interpretations of data a challenge. This fact likely explains
increased total lymphocyte count. LGL can be identified by morphol- the large differences in the frequency of specific clinical manifestations
13
ogy, although immunophenotyping is necessary to distinguish whether across LGLL studies and raises concerns about the validity of compar-
the LGLs are of T-cell or NK-cell lineage (Fig. 94–2). The median LGL ing outcome analyses from different centers. In the absence of molec-
count of patients with T-LGL leukemia is 4.2 × 10 /L (see Table 94–1). ular hallmarks, and with a significant level of overlap in clinical and
9
Most patients (70 percent) with T-LGLL have chronic neutropenia, laboratory features between reactive and neoplastic LGL proliferations,
and approximately 35 percent have neutrophil counts less than 0.5 × the diagnosis of LGLL hinges in part on the selection of the cut-off value
10 /L. Recurrent bacterial infections of the upper and lower respira- for blood LGL levels. Historically, a diagnosis of LGLL required the
13
9
tory tract are observed in 30 percent of patients. Opportunistic infec- detection of 2 × 10 /L or greater blood LGL. However, there are patients
9
tions, however, are uncommon. Anemia is observed in approximately with less than 2 × 10 /L blood LGL that clearly have monoclonal
9
50 percent of cases of T-LGLL. Transfusion-dependent anemia occurs CD8+ T-cell lymphoproliferations and display clinical and laboratory
in approximately 20 percent of patients. More than one mechanism of features, as well as outcomes, indistinguishable from LGLL. This finding
anemia has been described, including autoimmune hemolytic anemia has led to the adoption of less-restrictive criteria for the diagnosis of
(Chap. 54), pure red cell aplasia (Chap. 36), and, rarely, aplastic ane- LGLL. Currently, the most commonly accepted blood LGL cutoff for a
mia (Chap. 35). LGLL is the most commonly associated disease in diagnosis of LGLL is greater than 0.5 × 10 /L, with a TCRαβ+/CD3+/
9
3,43
patients with pure red cell aplasia. The role of LGL in the suppression CD8+/CD57+ immunophenotype, lasting more than 6 months. For
44
48
of erythropoiesis in a patient with LGLL was established in vitro and CLPD-NK (CD3–/CD8+/CD16+ and/or CD16+/CD56+) the cutoff is
45
the role of T-cells is supported by response to antithymocyte globulin. greater than 0.75 × 10 /L.
46
9
Thrombocytopenia, usually moderate, is seen in approximately 20 per-
cent of patients. It may be immune-mediated (presence of antiplatelet DIFFERENTIAL DIAGNOSIS
antibodies) (Chap. 117), a result of splenic sequestration, and rarely sec-
ondary to amegakaryocytosis. Rare cases of amegakaryocytic throm- The diagnosis of LGLL should be considered in patients with chronic
bocytopenia or red cell aplasia have occurred and have responded to or cyclic neutropenia and in patients with pure red cell aplasia or
49
immunosuppressive. 3,47 rheumatoid arthritis who have increased concentrations of LGL. HIV
Kaushansky_chapter 94_p1563-1568.indd 1566 9/18/15 10:53 AM