
Page 516 - Clinical Immunology_ Principles and Practice ( PDFDrive )
P. 516
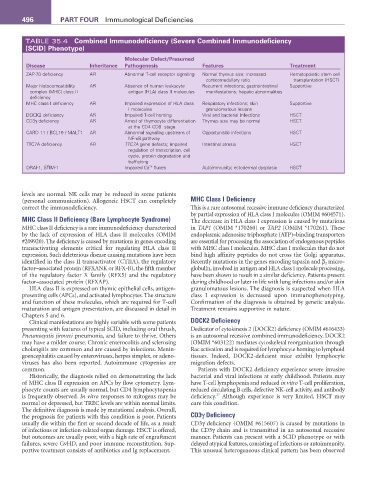
496 ParT fOur Immunological Deficiencies
TABLE 35.4 Combined Immunodeficiency (Severe Combined Immunodeficiency
[SCID] Phenotype)
Molecular Defect/Presumed
Disease Inheritance Pathogenesis features Treatment
ZAP-70 deficiency AR Abnormal T-cell receptor signaling Normal thymus size; increased Hematopoietic stem cell
corticomedullary ratio transplantation (HSCT)
Major histocompatibility AR Absence of human leukocyte Recurrent infections; gastrointestinal Supportive
complex (MHC) class II antigen (HLA) class II molecules manifestations; hepatic abnormalities
deficiency
MHC class I deficiency AR Impaired expression of HLA class Respiratory infections; skin Supportive
I molecules granulomatous lesions
DOCK2 deficiency AR Impaired T-cell homing Viral and bacterial infections HSCT
CD3γ deficiency AR Arrest of thymocyte differentiation Thymus size may be normal HSCT
at the CD4 CD8 stage
−
−
CARD 11 / BCL10 / MALT1 AR Abnormal signaling upstream of Opportunistic infections HSCT
NF-κB pathway
TTC7A deficiency AR TTC7A gene defects; impaired Intestinal atresia HSCT
regulation of transcription, cell
cycle, protein degradation and
trafficking
2+
ORAI-1, STIM-1 Impaired Ca fluxes Autoimmunity; ectodermal dysplasia HSCT
levels are normal. NK cells may be reduced in some patients
(personal communication). Allogeneic HSCT can completely MHC Class I Deficiency
correct the immunodeficiency. This is a rare autosomal recessive immune deficiency characterized
by partial expression of HLA class I molecules (OMIM #604571).
MHC Class II Deficiency (Bare Lymphocyte Syndrome) The decrease in HLA class I expression is caused by mutations
MHC class II deficiency is a rare immunodeficiency characterized in TAP1 (OMIM *170260) or TAP2 (OMIM *170261). These
by the lack of expression of HLA class II molecules (OMIM endoplasmic adenosine triphosphate (ATP)–binding transporters
#209920). The deficiency is caused by mutations in genes encoding are essential for processing the association of endogenous peptides
transactivating elements critical for regulating HLA class II with MHC class I molecules. MHC class I molecules that do not
expression. Such deleterious disease causing mutations have been bind high affinity peptides do not cross the Golgi apparatus.
identified in the class II transactivator (CTIIA), the regulatory Recently mutations in the genes encoding tapasin and β 2 micro-
factor–associated protein (RFXANK or RFX-B), the fifth member globulin, involved in antigen and HLA class I molecule processing,
of the regulatory factor X family (RFX5) and the regulatory have been shown to result in a similar deficiency. Patients present
factor–associated protein (RFXAP). during childhood or later in life with lung infections and/or skin
HLA class II is expressed on thymic epithelial cells, antigen- granulomatous lesions. The diagnosis is suspected when HLA
presenting cells (APCs), and activated lymphocytes. The structure class I expression is decreased upon immunophenotyping.
and function of these molecules, which are required for T-cell Confirmation of the diagnosis is obtained by genetic analysis.
maturation and antigen presentation, are discussed in detail in Treatment remains supportive in nature.
Chapters 5 and 6.
Clinical manifestations are highly variable with some patients DOCK2 Deficiency
presenting with features of typical SCID, including oral thrush, Dedicator of cytokinesis 2 (DOCK2) deficiency (OMIM #616433)
Pneumocystis jiroveci pneumonia, and failure to thrive. Others is an autosomal recessive combined immunodeficiency. DOCK2
may have a milder course. Chronic enterocolitis and sclerosing (OMIM *603122) mediates cytoskeletal reorganization through
cholangitis are common and are caused by infections. Menin- Rac activation and is required for lymphocyte homing to lymphoid
goencephalitis caused by enteroviruses, herpes simplex, or adeno- tissues. Indeed, DOCK2-deficient mice exhibit lymphocyte
viruses has also been reported. Autoimmune cytopenias are migration defects.
common. Patients with DOCK2 deficiency experience severe invasive
Historically, the diagnosis relied on demonstrating the lack bacterial and viral infections at early childhood. Patients may
of MHC class II expression on APCs by flow cytometry. Lym- have T-cell lymphopenia and reduced in vitro T-cell proliferation,
phocyte counts are usually normal, but CD4 lymphocytopenia reduced circulating B cells, defective NK-cell activity, and antibody
27
is frequently observed. In vitro responses to mitogens may be deficiency. Although experience is very limited, HSCT may
normal or depressed, but TREC levels are within normal limits. cure this condition.
The definitive diagnosis is made by mutational analysis. Overall,
the prognosis for patients with this condition is poor. Patients CD3γ Deficiency
usually die within the first or second decade of life, as a result CD3γ deficiency (OMIM #615607) is caused by mutations in
of infections or infection-related organ damage. HSCT is offered, the CD3γ chain and is transmitted in an autosomal recessive
but outcomes are usually poor, with a high rate of engraftment manner. Patients can present with a SCID phenotype or with
failures, severe GvHD, and poor immune reconstitution. Sup- delayed atypical features, consisting of infections or autoimmunity.
portive treatment consists of antibiotics and Ig replacement. This unusual heterogeneous clinical pattern has been observed