
Page 129 - First Aid for the USMLE Step 1 2020, Thirtieth edition [MedicalBooksVN.com]_Neat
P. 129
BIOCHEmISTRY ``BIOCHEMISTRY—METABOlISM BIOCHEmISTRY ``BIOCHEMISTRY—METABOlISM SECTION II 85
Cystinuria Hereditary defect of renal PCT and intestinal Autosomal recessive. Common (1:7000).
amino acid transporter that prevents Urinary cyanide-nitroprusside test is diagnostic.
A
reabsorption of Cystine, Ornithine, Lysine,
and Arginine (COLA).
Excess cystine in the urine can lead to recurrent Cystine is made of 2 cysteines connected by a
precipitation of hexagonal cystine stones A . disulfide bond.
Treatment: urinary alkalinization (eg, potassium
citrate, acetazolamide) and chelating agents
(eg, penicillamine) solubility of cystine
stones; good hydration.
Organic acidemias Most commonly present in infancy with poor feeding, vomiting, hypotonia, high anion gap
metabolic acidosis, hepatomegaly, seizures. Organic acid accumulation:
Inhibits gluconeogenesis fasting blood glucose levels, ketoacidosis high anion gap
metabolic acidosis
Inhibits urea cycle hyperammonemia
Propionic acidemia Deficiency of propionyl-CoA carboxylase Treatment: low-protein diet limited in
propionyl-CoA, methylmalonic acid. substances that metabolize into propionyl-
CoA: Valine, Odd-chain fatty acids,
Methylmalonic Deficiency of methylmalonyl-CoA mutase or Methionine, Isoleucine, Threonine
acidemia vitamin B 12 . (VOMIT).
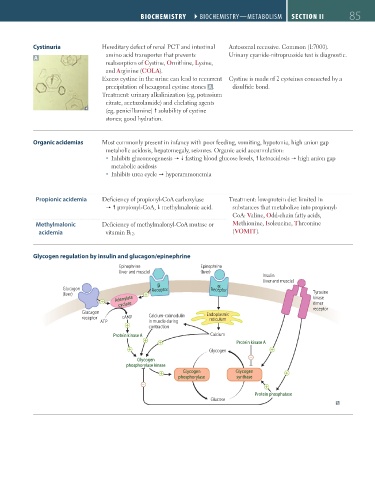
Glycogen regulation by insulin and glucagon/epinephrine
Epinephrine Epinephrine
(liver and muscle) (liver)
Insulin
(liver and muscle)
Glucagon Receptor Receptor
(liver) Tyrosine
Adenylate kinase
cyclase dimer
receptor
Glucagon Endoplasmic
receptor cAMP Calcium-calmodulin reticulum
ATP in muscle during
contraction
Protein kinase A Calcium
Protein kinase A
Glycogen
−
Glycogen
phosphorylase kinase
Glycogen Glycogen
phosphorylase synthase
−
Protein phosphatase
Glucose
FAS1_2019_01-Biochem.indd 85 11/7/19 3:16 PM