
Page 594 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 594
Chapter 38 Heme Biosynthesis and Its Disorders 509
sideroblasts. TRMA presents with early-onset megaloblastic anemia,
Therapy for Hereditary Sideroblastic Anemia
diabetes mellitus, and sensorineural deafness, which respond variably
A trial of pyridoxine (100–200 mg/day taken orally) is indicated for 3 to thiamine treatment.
months for all patients with hereditary sideroblastic anemia. Response Missense and nonsense mutations in the PUS1 gene coding for
is variable and ranges from complete correction of hemoglobin levels pseudouridine synthase-1 cause the rare autosomal recessive disease,
to no effect. Even when pyridoxine completely corrects the anemia myopathy, lactic acidosis, and sideroblastic anemia (MLASA). 192–195
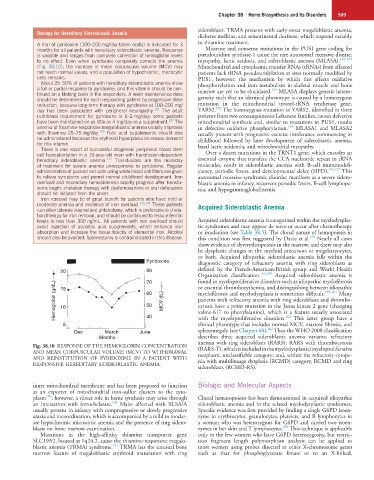
(Fig. 38.10), the increase in mean corpuscular volume (MCV) may Mitochondrial and cytoplasmic transfer RNAs (tRNAs) from affected
not reach normal values, and a population of hypochromic, microcytic patients lack tRNA pseudouridylation at sites normally modified by
cells remains. PUS1; however, the mechanism by which this affects oxidative
About 25–50% of patients with hereditary sideroblastic anemia show phosphorylation and iron metabolism in skeletal muscle and bone
a full or partial response to pyridoxine, and this vitamin should be con- marrow are yet to be elucidated. MLASA displays genetic hetero-
193
tinued on a lifelong basis in the responders. A lower maintenance dose
should be determined for each responding patient by progressive dose geneity such that an identical phenotype is caused by a homozygous
reduction, because long-term therapy with pyridoxine at 100–200 mg/ mutation in the mitochondrial tyrosyl-tRNA synthetase gene,
194
day has been associated with peripheral neuropathy. 175 The adult YARS2. The homozygous mutation in YARS2, identified in three
nutritional requirement for pyridoxine is 1–2 mg/day; some patients patients from two consanguineous Lebanese families, causes defective
have been maintained on as little as 4 mg/day as a supplement. 157 The mitochondrial synthesis and, similar to mutations in PUS1, results
194
anemia of thiamine-responsive megaloblastic anemia usually improves in defective oxidative phosphorylation. MLASA1 and MLASA2
with thiamine 25–75 mg/day. 176 Folic acid supplements should also usually present with progressive exercise intolerance commencing in
be administered because the erythroid hyperplasia increases demand childhood followed by later development of sideroblastic anemia,
for this vitamin. basal lactic acidemia, and mitochondrial myopathy.
There is one report of successful allogeneic peripheral blood stem
cell transplantation in a 19-year-old man with transfusion-dependent Over a dozen mutations in the TRNT1 gene, which encodes an
hereditary sideroblastic anemia. 177 Transfusions are the mainstay essential enzyme that transfers the CCA nucleotide repeat to tRNA
of treatment for severe anemia unresponsive to pyridoxine. Regular molecules, result in sideroblastic anemia with B-cell immunodefi-
administration of packed red cells using white blood cell filters are given ciency, periodic fevers, and developmental delay (SIFD). 196,197 This
to relieve symptoms and permit normal childhood development. Iron autosomal recessive syndromic disorder manifests as a severe sidero-
overload and secondary hemosiderosis rapidly progress after transfu- blastic anemia in infancy, recurrent periodic fevers, B-cell lymphope-
sions begin; chelation therapy with desferrioxamine or oral deferasirox nia, and hypogammaglobulinemia.
should be initiated from the onset.
Iron removal may be of great benefit for patients who have mild or
moderate anemia and evidence of iron overload. 172,173 These patients Acquired Sideroblastic Anemia
can often tolerate intermittent phlebotomy, which is preferable to chela-
tion therapy for iron removal, and should be continued to reduce ferritin
levels to less than 300 ng/mL. All patients with iron overload should Acquired sideroblastic anemia is categorized within the myelodysplas-
avoid ingestion of ascorbic acid supplements, which enhance iron tic syndromes and may appear de novo or occur after chemotherapy
absorption and increase the tissue toxicity of elemental iron. Alcohol or irradiation (see Table 38.5). The clonal nature of hemopoiesis in
153
should also be avoided. Splenectomy is contraindicated in this disease. this condition was first suggested by Dacie et al. Nearly all cases
show evidence of dyserythropoiesis in the marrow, and there may also
be dysplastic changes in the myeloid precursors or megakaryocytes,
or both. Acquired idiopathic sideroblastic anemia falls within the
Pyridoxine diagnostic category of refractory anemia with ring sideroblasts as
20 80 defined by the French-American-British group and World Health
198,199
Organization classification. Acquired sideroblastic anemia is
found in myeloproliferative disorders such as idiopathic myelofibrosis
70
Hemoglobin (g/dL) 15 60 MCV (fL) myelofibrosis and myelodysplasia is sometimes difficult. 199–201 Many
or essential thrombocythemia, and distinguishing between idiopathic
patients with refractory anemia with ring sideroblasts and thrombo-
50
cytosis have a point mutation in the Janus kinase 2 gene (changing
10
valine-617 to phenylalanine), which is a feature usually associated
202
clinical phenotype that includes normal MCV, marrow fibrosis, and
5 40 with the myeloproliferative disorders. This latter group have a
202
Dec March June splenomegaly (see Chapter 60). Thus the WHO 2008 classification
Months describes three acquired sideroblastic anemia variants: refractory
anemia with ring sideroblasts (RARS); RARS with thrombocytosis
Fig. 38.10 RESPONSE OF THE HEMOGLOBIN CONCENTRATION (RARS-T), which is included in the myelodysplastic/myeloproliferative
AND MEAN CORPUSCULAR VOLUME (MCV) TO WITHDRAWAL neoplasm, unclassifiable category; and, within the refractory cytope-
AND REINSTITUTION OF PYRIDOXINE IN A PATIENT WITH nia with multilineage dysplasia (RCMD) category, RCMD and ring
RESPONSIVE HEREDITARY SIDEROBLASTIC ANEMIA.
sideroblasts (RCMD-RS).
inner mitochondrial membrane and has been proposed to function Biologic and Molecular Aspects
as an exporter of mitochondrial iron-sulfur clusters to the cyto-
189
plasm ; however, a direct role in heme synthesis may arise through Clonal hematopoiesis has been demonstrated in acquired idiopathic
190
an interaction with ferrochelatase. Males affected with XLSA/A sideroblastic anemia and in the related myelodysplastic syndromes.
usually present in infancy with nonprogressive or slowly progressive Specific evidence was first provided by finding a single G6PD isoen-
ataxia and incoordination, which is accompanied by a mild to moder- zyme in erythrocytes, granulocytes, platelets, and B lymphocytes in
ate hypochromic microcytic anemia and the presence of ring sidero- a woman who was heterozygous for G6PD and carried two isoen-
203
blasts on bone marrow examination. zymes in her skin and T lymphocytes. This technique is applicable
Mutations in the high-affinity thiamine transporter gene only to the few women who have G6PD heterozygosity, but restric-
SLC19A2, located at 1q24.2, cause the thiamine-responsive megalo- tion fragment length polymorphism analysis can be applied to
191
blastic anemia (TRMA) syndrome. TRMA has the unusual bone most women using probes directed at other X-chromosome genes
marrow feature of megaloblastic erythroid maturation with ring such as that for phosphoglycerate kinase or to an X-linked,