
Page 592 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 592
Chapter 38 Heme Biosynthesis and Its Disorders 507
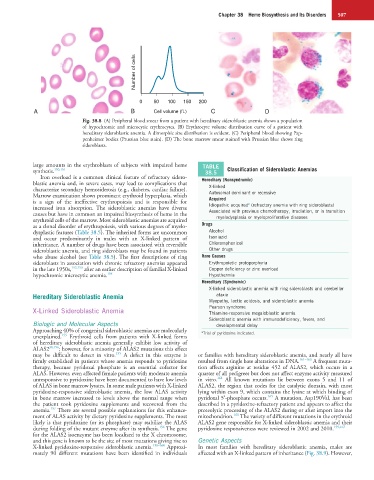
Number of cells
0 50 100 150 200
A B Cell volume (fL) C D
Fig. 38.8 (A) Peripheral blood smear from a patient with hereditary sideroblastic anemia shows a population
of hypochromic and microcytic erythrocytes. (B) Erythrocyte volume distribution curve of a patient with
hereditary sideroblastic anemia. A dimorphic size distribution is evident. (C) Peripheral blood showing Pap-
penheimer bodies (Prussian blue stain). (D) The bone marrow smear stained with Prussian blue shows ring
sideroblasts.
large amounts in the erythroblasts of subjects with impaired heme TABLE
synthesis. 150,151 38.5 Classification of Sideroblastic Anemias
Iron overload is a common clinical feature of refractory sidero-
blastic anemia and, in severe cases, may lead to complications that Hereditary (Nonsyndromic)
characterize secondary hemosiderosis (e.g., diabetes, cardiac failure). X-linked
Marrow examination shows prominent erythroid hyperplasia, which Autosomal dominant or recessive
is a sign of the ineffective erythropoiesis and is responsible for Acquired a
increased iron absorption. The sideroblastic anemias have diverse Idiopathic acquired (refractory anemia with ring sideroblasts)
causes but have in common an impaired biosynthesis of heme in the Associated with previous chemotherapy, irradiation, or in transition
erythroid cells of the marrow. Most sideroblastic anemias are acquired myelodysplasia or myeloproliferative diseases
as a clonal disorder of erythropoiesis, with various degrees of myelo- Drugs
dysplastic features (Table 38.5). The inherited forms are uncommon Alcohol
and occur predominantly in males with an X-linked pattern of Isoniazid
inheritance. A number of drugs have been associated with reversible Chloramphenicol
sideroblastic anemia, and ring sideroblasts may be found in patients Other drugs
who abuse alcohol (see Table 38.5). The first descriptions of ring Rare Causes
sideroblasts in association with chronic refractory anemias appeared Erythropoietic protoporphyria
in the late 1950s, 152,153 after an earlier description of familial X-linked Copper deficiency or zinc overload
hypochromic microcytic anemia. 154 Hypothermia
Hereditary (Syndromic)
X-linked sideroblastic anemia with ring sideroblasts and cerebellar
Hereditary Sideroblastic Anemia ataxia
Myopathy, lactic acidosis, and sideroblastic anemia
Pearson syndrome
X-Linked Sideroblastic Anemia Thiamine-responsive megaloblastic anemia
Sideroblastic anemia with immunodeficiency, fevers, and
Biologic and Molecular Aspects developmental delay
Approaching 40% of congenital sideroblastic anemias are molecularly a Trial of pyridoxine indicated.
155
unexplained. Erythroid cells from patients with X-linked forms
of hereditary sideroblastic anemia generally exhibit low activity of
ALAS2 30,156 ; however, for a minority of ALAS2 mutations this effect
155
may be difficult to detect in vitro. A defect in this enzyme is or families with hereditary sideroblastic anemia, and nearly all have
firmly established in patients whose anemia responds to pyridoxine resulted from single base alterations in DNA. 161–163 A frequent muta-
therapy, because pyridoxal phosphate is an essential cofactor for tion affects arginine at residue 452 of ALAS2, which occurs in a
ALAS. However, even affected female patients with moderate anemia quarter of all pedigrees but does not affect enzyme activity measured
164
unresponsive to pyridoxine have been documented to have low levels in vitro. All known mutations lie between exons 5 and 11 of
of ALAS in bone marrow lysates. In some male patients with X-linked ALAS2, the region that codes for the catalytic domain, with most
pyridoxine-responsive sideroblastic anemia, the low ALAS activity lying within exon 9, which contains the lysine at which binding of
165
in bone marrow increased to levels above the normal range when pyridoxal 5′-phosphate occurs. A mutation, Asp190Val, has been
the patient took pyridoxine supplements and recovered from the described in a pyridoxine-refractory patient and appears to affect the
157
anemia. There are several possible explanations for this enhance- proteolytic processing of the ALAS2 during or after import into the
166
ment of ALAS activity by dietary pyridoxine supplements. The most mitochondrion. The variety of different mutations in the erythroid
likely is that pyridoxine (or its phosphate) may stabilize the ALAS ALAS2 gene responsible for X-linked sideroblastic anemia and their
156
during folding of the mutant enzyme after its synthesis. The gene pyridoxine responsiveness were reviewed in 2002 and 2010. 159,167
for the ALAS2 isoenzyme has been localized to the X chromosome,
and this gene is known to be the site of most mutations giving rise to Genetic Aspects
X-linked pyridoxine-responsive sideroblastic anemia. 158–160 Approxi- In most families with hereditary sideroblastic anemia, males are
mately 90 different mutations have been identified in individuals affected with an X-linked pattern of inheritance (Fig. 38.9). However,