
Page 663 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 663
Chapter 40 Thalassemia Syndromes 565
TABLE Transfusion-Dependent Thalassemia Screening Recommendations
40.5
MRI with T2* liver iron content Liver iron every 1–2 years
MRI with T2* cardiac iron content Cardiac iron every 1–2 years
Ferritin Total body iron every 3 months
History and physical exam General health, medication compliance every 3–4 months
Echocardiogram Pulmonary hypertension TRV every 1–2 years
Liver function panel Liver failure, hepatitis every 3 months
Liver ultrasound (if LIC>5/ferritin >800) Cirrhosis screening annually
AFP (if >40 or presence of clinical cirrhosis) Hepatocellular carcinoma screening annually
Hepatitis B, C serologies (if receiving blood transfusions) Hepatitis B and C viral infection/exposure annually
Tanner stage/Sexual development evaluation Sexual development annually
Standing and sitting height Growth and development every 6 months
Free T4, TSH Thyroid function annually
Calcium, phosphate, vitamin D Parathyroid function annually
Fasting blood sugar/oral glucose tolerance test Diabetes mellitus screening annually
ACTH test Adrenal insufficiency screening annually
DEXA Bone mineral density annually
NTX, CTX, AP Bone mineral density annually
Dental evaluation Maxillofacial disease, periodontal disease, caries 6–12 months
ACTH, Adrenocorticotropic hormone; AFP, α-fetoprotein; AP, alkaline phosphatase; CTX, collagen type 1 cross-linked C-telopeptide; DEXA, dual-energy x-ray
absorptiometry; LIC, liver iron concentration; MRI, magnetic resonance imaging; NTX, N-terminal telopeptide; TRV, tricuspic regurgitant velocity; TSH, thyroid stimulating
hormone.
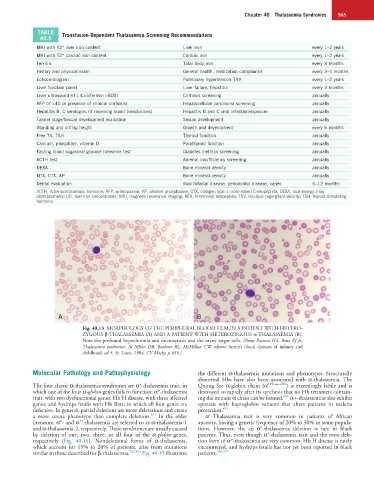
A B
Fig. 40.13 MORPHOLOGY OF THE PERIPHERAL BLOOD FILM IN A PATIENT WITH HETERO-
ZYGOUS β-THALASSEMIA (A) AND A PATIENT WITH HETEROZYGOUS α-THALASSEMIA (B).
Note the profound hypochromia and microcytosis and the many target cells. (From Pearson HA, Benz EJ Jr:
Thalassemia syndromes. In Miller DR, Baehner RL, McMillan CW, editors: Smith’s blood diseases of infancy and
childhood, ed 5, St. Louis, 1984, CV Mosby, p 439.)
Molecular Pathology and Pathophysiology the different α-thalassemia mutations and phenotypes. Structurally
abnormal Hbs have also been associated with α-thalassemia. The
+
The four classic α-thalassemia syndromes are α -thalassemia trait, in Quong Sze α-globin chain (α 125Leu→Pro ) is exceedingly labile and is
which one of the four α-globin genes fails to function; α°-thalassemia destroyed so rapidly after its synthesis that no Hb tetramers contain-
454
trait, with two dysfunctional genes; Hb H disease, with three affected ing the mutant α chain can be formed. α+-thalassemias also exhibit
genes; and hydrops fetalis with Hb Bart, in which all four genes are epistasis with haptoglobin variants that alters patterns in malaria
defective. In general, partial deletions are more deleterious and create protection. 455
+
451
a more severe phenotype than complete deletions. In the older α -Thalassemia trait is very common in patients of African
++
literature, α°- and α -thalassemia are referred to as α-thalassemia-1 ancestry, having a genetic frequency of 20% to 30% in some popula-
and α-thalassemia-2, respectively. These syndromes are usually caused tions. However, the cis α°-thalassemia deletion is rare in black
+
by deletion of one, two, three, or all four of the α-globin genes, patients. Thus, even though α -thalassemia trait and the trans dele-
respectively (Fig. 40.14). Nondeletional forms of α-thalassemia, tion form of α°-thalassemia are very common, Hb H disease is rarely
which account for 15% to 20% of patients, arise from mutations encountered, and hydrops fetalis has not yet been reported in black
similar to those described for β-thalassemia. 452,453 Fig. 40.15 illustrates patients. 456,457