
Page 2000 - Williams Hematology ( PDFDrive )
P. 2000
1974 Part XII: Hemostasis and Thrombosis Chapter 115: Vascular Function In Hemostasis 1975
impaired healing of cutaneous wounds, a response that appears to restenosis (Fig. 115–7C and Table 115–3). This process reflects leu-
169
depend largely on the fibrinolytic action of plasmin as loss of fibrinogen kocyte invasion, proliferation and migration of smooth muscle cells,
eliminates these defects. Mice doubly deficient in plasminogen and deposition of extracellular matrix, and reendothelialization. Electrical
170
apolipoprotein E (ApoE) showed an increased predisposition to athero- or mechanical injury studies in gene-targeted mice indicate that neo-
171
sclerosis compared to animals deficient in ApoE alone (Fig. 115–7A). intima formation, an initial step in restenosis, requires intact expres-
Mice with ApoE deficiency combined with deficiency of either u-PA or sion of plasminogen and u-PA, but not t-PA. 180–182 Interestingly, loss of
183
t-PA showed the same predilection for early fatty streaks and advanced uPAR has no effect on neointima formation, whereas loss of PAI-1 is
plaques as was observed in mice with isolated ApoE deficiency, suggest- associated with increased neointimal stenosis. 184,185 In these injury mod-
ing that complete elimination of plasmin generating activity is required els, which do not induce severe thrombosis, it is thought that vascular
172
to exacerbate the proatherogenic state. Finally, mice doubly deficient occlusion, reflecting migration of smooth muscle cells and leukocytes,
in ApoE and PAI-1 exhibit no change in early plaque size at the aortic is impaired when fibrinolytic potential is attenuated. 186
root, 173,174 decreased early plaque size at the carotid bifurcation, 173,174 but In the ferric chloride, Rose Bengal, and copper cuff models, on the
increased advanced plaque size with accelerated deposition of matrix. 175 other hand, thrombosis is observed within minutes of arterial injury
Once the atherosclerotic plaque is established, plasmin may affect (see Fig. 115–7 and Table 115–3). In these systems, deficiency of PAI-1
its evolution by mediating invasion of leukocytes (see Table 115–3). is associated with later and less-extensive thrombotic occlusion of the
176
In the peritoneal cavity, recruitment of inflammatory cells is profoundly injured artery, 187,188 while loss of u-PA is associated with more rapid and
177
influenced by the presence or absence of plasminogen. In trans- more significant thrombotic occlusion. At the same time, the absence
189
plant-associated arteriosclerosis, the extent of disease is significantly of PAI-1 led to reduced vascular stenosis, regardless of whether ApoE
reduced in plasminogen-deficient mice, reflecting, at least in part, was absent 190,191 or present. 192,193 In balloon-injured rat carotid arter-
reduced influx of macrophages, with an associated reduction in medial ies, finally, transduction of a PAI-1–expressing gene led to increased
necrosis, fragmentation of elastic laminae, and remodeling of the adven- restenosis of the vessel, again suggesting that clearance of the initial
178
titia. Thus, the role of plasmin in degrading fibrin and other matrix thrombus may have longterm effects on vessel patency and neointima
constituents in the early lesion limits atherosclerosis, whereas its ability formation. In these models, the predominant effect of the fibrinolytic
194
to promote cellular invasion later on appears to promote atherogenesis. system may be to clear the initial thrombus, which may provide a provi-
During aortic aneurysm formation in mice, deficiency of u-PA, but sional scaffolding for later restenosis.
not t-PA, was associated with reduced medial destruction and impaired
activation of downstream plasmin-dependent matrix metalloprotein-
ases (Fig. 115–7B and Table 115–3). Similarly, u-PA–, but not t-PA–,
172
deficient mice were protected from cardiac rupture secondary to ven- FIBRINOLYTIC ASSEMBLY AND
tricular aneurysm. In this study, temporary administration of PAI-1 or VASCULAR DISEASE
the general matrix metalloproteinase inhibitor, tissue inhibitor of metal-
loproteinase (TIMP)-1, completely protected wild-type mice from aor- Endothelial cells use receptors, primarily uPAR and the annexin A2/
tic rupture, reinforcing the concept that plasmin-based protease activity S100A10 system, to assemble the fibrinolytic system on their sur-
promotes aneurysm progression. 179 face (Chap. 135; Fig. 115–6). Recent evidence suggests that impair-
Vascular remodeling may occur following acute arterial injury ment of receptor-mediated fibrinolytic assembly may lead to vascular
induced by interventions for vascular compromise, leading to vascular compromise.
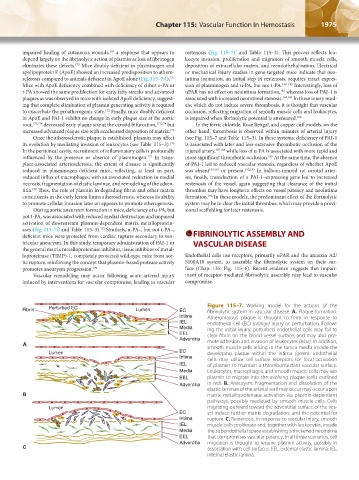
Fibrin Perturbed EC Lumen EC Figure 115–7. Working model for the actions of the
fibrinolytic system in vascular disease. A. Plaque formation.
Intima Atheromatous plaque is thought to form in response to
IEL endothelial cell (EC) (orange) injury or perturbation. Follow-
Media ing the initial injury, perturbed endothelial cells may fail to
EEL clear fibrin on the blood vessel surface, and may also pro-
Adventitia
A mote adhesion and invasion of leukocytes (blue). In addition,
smooth muscle cells arising in the tunica media invade the
Lumen EC developing plaque within the intima (green). Endothelial
Intima cells may utilize cell-surface receptors for focal activation
IEL of plasmin to maintain a thromboresistant vascular surface.
Media Leukocytes, macrophages, and smooth muscle cells may use
EEL plasmin to migrate into the evolving plaque (cells outlined
Adventitia in red). B. Aneurysm. Fragmentation and dissolution of the
elastic laminae of the arterial wall may occur may occur upon
B matrix metalloproteinase activation via plasmin-dependent
pathways, possibly mediated by smooth muscle cells. Cells
Lumen migrating outward toward the adventitial surface of the ves-
EC sel induce further matrix degradation, and the potential for
Intima rupture. C. Restenosis. In response to vascular injury, smooth
IEL muscle cells proliferate and, together with leukocytes, invade
Media the subendothelial space establishing a thickened neointima
EEL that compromises vascular patency. In all three scenarios, cell
Adventitia migration is thought to require plasmin activity, possibly in
C association with cell surfaces. EEL, external elastic lamina; IEL,
internal elastic lamina.
Kaushansky_chapter 115_p1967-1984.indd 1975 9/18/15 10:08 AM