
Page 251 - The Netter Collection of Medical Illustrations - Integumentary System_ Volume 4 ( PDFDrive )
P. 251
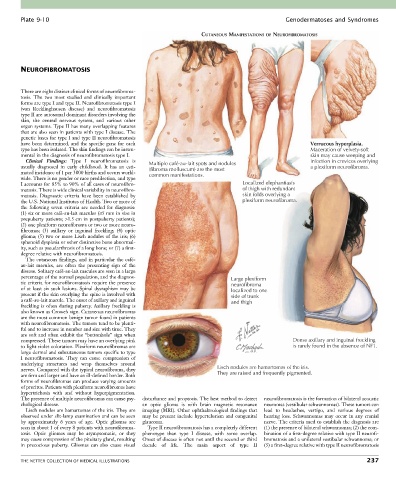
Plate 9-10 Genodermatoses and Syndromes
CUTANEOUS MANIFESTATIONS OF NEUROFIBROMATOSIS
NEUROFIBROMATOSIS
There are eight distinct clinical forms of neurofibroma-
tosis. The two most studied and clinically important
forms are type I and type II. Neurofibromatosis type I
(von Recklinghausen disease) and neurofibromatosis
type II are autosomal dominant disorders involving the
skin, the central nervous system, and various other
organ systems. Type II has many overlapping features
that are also seen in patients with type I disease. The
genetic bases for type I and type II neurofibromatosis
have been determined, and the specific gene for each Verrucous hyperplasia.
type has been isolated. The skin findings can be instru- Maceration of velvety-soft
mental in the diagnosis of neurofibromatosis type I. skin may cause weeping and
Clinical Findings: Type I neurofibromatosis is infection in crevices overlying
usually diagnosed in early childhood. It has an esti- Multiple café-au-lait spots and nodules a plexiform neurofibroma.
(fibroma molluscum) are the most
mated incidence of 1 per 3000 births and occurs world- common manifestations.
wide. There is no gender or race predilection, and type
I accounts for 85% to 90% of all cases of neurofibro- Localized elephantiasis
matosis. There is wide clinical variability in neurofibro- of thigh with redundant
matosis. Diagnostic criteria have been established by skin folds overlying a
the U.S. National Institutes of Health. Two or more of plexiform neurofibroma.
the following seven criteria are needed for diagnosis:
(1) six or more café-au-lait macules (≥5 mm in size in
prepuberty patients; >1.5 cm in postpuberty patients);
(2) one plexiform neurofibroma or two or more neuro-
fibromas; (3) axillary or inguinal freckling; (4) optic
glioma; (5) two or more Lisch nodules of the iris; (6)
sphenoid dysplasia or other distinctive bone abnormal-
ity, such as pseudarthrosis of a long bone; or (7) a first-
degree relative with neurofibromatosis.
The cutaneous findings, and in particular the café-
au-lait macules, are often the presenting sign of the
disease. Solitary café-au-lait macules are seen in a large
percentage of the normal population, and the diagnos- Large plexiform
tic criteria for neurofibromatosis require the presence neurofibroma
of at least six such lesions. Spinal dysraphism may be localized to one
present if the skin overlying the spine is involved with side of trunk
a café-au-lait macule. The onset of axillary and inguinal and thigh
freckling is often during puberty. Axillary freckling is
also known as Crowe’s sign. Cutaneous neurofibromas
are the most common benign tumor found in patients
with neurofibromatosis. The tumors tend to be plenti-
ful and to increase in number and size with time. They
are soft and often exhibit the “buttonhole” sign when
compressed. These tumors may have an overlying pink Dense axillary and inguinal freckling
to light violet coloration. Plexiform neurofibromas are is rarely found in the absence of NF1.
large dermal and subcutaneous tumors specific to type
I neurofibromatosis. They can cause compression of
underlying structures and wrap themselves around
nerves. Compared with the typical neurofibroma, they Lisch nodules are hamartomas of the iris.
are firm and larger and have an ill-defined border. Both They are raised and frequently pigmented.
forms of neurofibromas can produce varying amounts
of pruritus. Patients with plexiform neurofibromas have
hypertrichosis with and without hyperpigmentation.
The presence of multiple neurofibromas can cause psy- disturbance and proptosis. The best method to detect neurofibromatosis is the formation of bilateral acoustic
chological disease. an optic glioma is with brain magnetic resonance neuromas (vestibular schwannomas). These tumors can
Lisch nodules are hamartomas of the iris. They are imaging (MRI). Other ophthalmological findings that lead to headaches, vertigo, and various degrees of
observed under slit-lamp examination and can be seen may be present include hypertelorism and congenital hearing loss. Schwannomas may occur in any cranial
by approximately 6 years of age. Optic gliomas are glaucoma. nerve. The criteria used to establish the diagnosis are
seen in about 1 of every 8 patients with neurofibroma- Type II neurofibromatosis has a completely different (1) the presence of bilateral schwannomas; (2) the com-
tosis. Optic gliomas may be asymptomatic, or they phenotype than type I disease, with some overlap. bination of a first-degree relative with type II neurofi-
may cause compression of the pituitary gland, resulting Onset of disease is often not until the second or third bromatosis and a unilateral vestibular schwannoma; or
in precocious puberty. Gliomas can also cause visual decade of life. The main aspect of type II (3) a first-degree relative with type II neurofibromatosis
THE NETTER COLLECTION OF MEDICAL ILLUSTRATIONS 237