
Page 845 - Hematology_ Basic Principles and Practice ( PDFDrive )
P. 845
728 Part VI Non-Malignant Leukocytes
systemic disease and involvement of the orbit and skull. Fewer than syndrome. This CNS involvement is typically seen in children who
one-third of children who ultimately develop DI have polydipsia and had classic lytic bony involvement or soft tissue mass lesions in or
polyuria as presenting symptoms of LCH. Most cases of DI present around the CNS years earlier. Delayed CNS involvement is typically
within 4 years of diagnosis. DI is caused by infiltration by Langerhans diagnosed after a prolonged, sometimes insidious, decrease in school
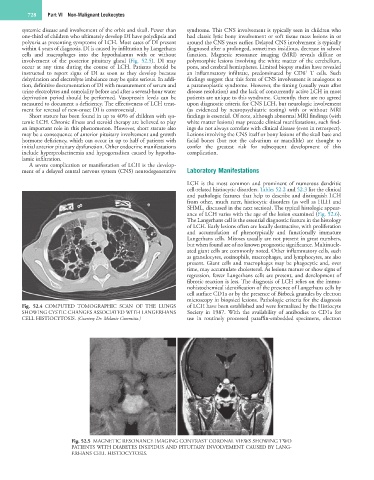
cells and macrophages into the hypothalamus with or without function. Magnetic resonance imaging (MRI) reveals diffuse or
involvement of the posterior pituitary gland (Fig. 52.5). DI may polymorphic lesions involving the white matter of the cerebellum,
occur at any time during the course of LCH. Patients should be pons, and cerebral hemispheres. Limited biopsy studies have revealed
+
instructed to report signs of DI as soon as they develop because an inflammatory infiltrate, predominated by CD8 T cells. Such
dehydration and electrolyte imbalance may be quite serious. In addi- findings suggest that this form of CNS involvement is analogous to
tion, definitive documentation of DI with measurement of serum and a paraneoplastic syndrome. However, the timing (usually years after
urine electrolytes and osmolality before and after a several-hour water disease resolution) and the lack of concurrently active LCH in most
deprivation period should be performed. Vasopressin levels can be patients are unique to this syndrome. Currently, there are no agreed
measured to document a deficiency. The effectiveness of LCH treat- upon diagnostic criteria for CNS LCH, but neurologic involvement
ment for reversal of new-onset DI is controversial. (as evidenced by neuropsychiatric testing) with or without MRI
Short stature has been found in up to 40% of children with sys- findings is essential. Of note, although abnormal MRI findings (with
temic LCH. Chronic illness and steroid therapy are believed to play white matter lesions) may precede clinical manifestations, such find-
an important role in this phenomenon. However, short stature also ings do not always correlate with clinical disease (even in retrospect).
may be a consequence of anterior pituitary involvement and growth Lesions involving the CNS itself or bony lesions of the skull base and
hormone deficiency, which can occur in up to half of patients with facial bones (but not the calvarium or mandible) are thought to
initial anterior pituitary dysfunction. Other endocrine manifestations confer the greatest risk for subsequent development of this
include hyperprolactinemia and hypogonadism caused by hypotha- complication.
lamic infiltration.
A severe complication or manifestation of LCH is the develop-
ment of a delayed central nervous system (CNS) neurodegenerative Laboratory Manifestations
LCH is the most common and prominent of numerous dendritic
cell-related histiocytic disorders. Tables 52.2 and 52.3 list the clinical
and pathologic features that help to describe and distinguish LCH
from other, much rarer, histiocytic disorders (as well as HLH and
SHML, discussed in the next section). The typical histologic appear-
ance of LCH varies with the age of the lesion examined (Fig. 52.6).
The Langerhans cell is the essential diagnostic feature in the histology
of LCH. Early lesions often are locally destructive, with proliferation
and accumulation of phenotypically and functionally immature
Langerhans cells. Mitoses usually are not present in great numbers,
but when found are of no known prognostic significance. Multinucle-
ated giant cells are commonly noted. Other inflammatory cells, such
as granulocytes, eosinophils, macrophages, and lymphocytes, are also
present. Giant cells and macrophages may be phagocytic and, over
time, may accumulate cholesterol. As lesions mature or show signs of
regression, fewer Langerhans cells are present, and development of
fibrotic reaction is less. The diagnosis of LCH relies on the immu-
nohistochemical identification of the presence of Langerhans cells by
cell surface CD1a or by the presence of Birbeck granules by electron
microscopy in biopsied lesions. Pathologic criteria for the diagnosis
Fig. 52.4 COMPUTED TOMOGRAPHIC SCAN OF THE LUNGS of LCH have been established and were formalized by the Histiocyte
SHOWING CYSTIC CHANGES ASSOCIATED WITH LANGERHANS Society in 1987. With the availability of antibodies to CD1a for
CELL HISTIOCYTOSIS. (Courtesy Dr. Melanie Committo.) use in routinely processed paraffin-embedded specimens, electron
Fig. 52.5 MAGNETIC RESONANCE IMAGING CONTRAST CORONAL VIEWS SHOWING TWO
PATIENTS WITH DIABETES INSIPIDUS AND PITUITARY INVOLVEMENT CAUSED BY LANG-
ERHANS CELL HISTIOCYTOSIS.