
Page 855 - Clinical Immunology_ Principles and Practice ( PDFDrive )
P. 855
826 ParT SIX Systemic Immune Diseases
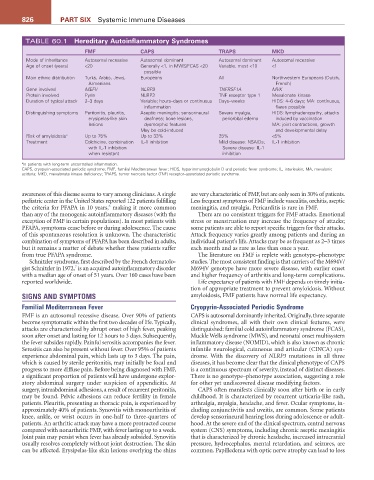
TABLE 60.1 Hereditary autoinflammatory Syndromes
FMF CaPS TraPS MKD
Mode of inheritance Autosomal recessive Autosomal dominant Autosomal dominant Autosomal recessive
Age of onset (years) <20 Generally <1, in MWS/FCAS <20 Variable, most <10 <1
possible
Main ethnic distribution Turks, Arabs, Jews, Europeans All Northwestern Europeans (Dutch,
Armenians French)
Gene involved MEFV NLRP3 TNFRSF1A MVK
Protein involved Pyrin NLRP3 TNF receptor type 1 Mevalonate kinase
Duration of typical attack 2–3 days Variable; hours–days or continuous Days–weeks HIDS: 4–6 days; MA: continuous,
inflammation flares possible
Distinguishing symptoms Peritonitis, pleuritis, Aseptic meningitis; sensorineural Severe myalgia, HIDS: lymphadenopathy, attacks
erysipelas-like skin deafness; bone lesions, periorbital edema induced by vaccination
lesions dysmorphic features MA: joint contractions, growth
May be cold-induced and developmental delay
Risk of amyloidosis a Up to 75% Up to 33% 25% <5%
Treatment Colchicine, combination IL-1 inhibition Mild disease: NSAIDs; IL-1 inhibition
with IL-1 inhibition Severe disease: IL-1
when resistant inhibition
a In patients with long-term uncontrolled inflammation.
CAPS, cryopyrin-associated periodic syndrome; FMF, familial Mediterranean fever; HIDS, hyperimmunoglobulin D and periodic fever syndrome; IL, interleukin; MA, mevalonic
aciduria; MKD, mevalonate kinase deficiency; TRAPS, tumor necrosis factor (TNF) receptor–associated periodic syndrome.
awareness of this disease seems to vary among clinicians. A single are very characteristic of FMF, but are only seen in 30% of patients.
pediatric center in the United States reported 122 patients fulfilling Less frequent symptoms of FMF include vasculitis, orchitis, aseptic
6
the criteria for PFAPA in 10 years, making it more common meningitis, and myalgia. Pericarditis is rare in FMF.
than any of the monogenic autoinflammatory diseases (with the There are no consistent triggers for FMF attacks. Emotional
exception of FMF in certain populations). In most patients with stress or menstruation may increase the frequency of attacks;
PFAPA, symptoms cease before or during adolescence. The cause some patients are able to report specific triggers for their attacks.
of this spontaneous resolution is unknown. The characteristic Attack frequency varies greatly among patients and during an
combination of symptoms of PFAPA has been described in adults, individual patient’s life. Attacks may be as frequent as 2–3 times
but it remains a matter of debate whether these patients suffer each month and as rare as less than once a year.
from true PFAPA syndrome. The literature on FMF is replete with genotype–phenotype
Schnitzler syndrome, first described by the French dermatolo- studies. The most consistent finding is that carriers of the M694V/
7
gist Schnitzler in 1972, is an acquired autoinflammatory disorder M694V genotype have more severe disease, with earlier onset
with a median age of onset of 51 years. Over 160 cases have been and higher frequency of arthritis and long-term complications.
reported worldwide. Life expectancy of patients with FMF depends on timely initia-
tion of appropriate treatment to prevent amyloidosis. Without
SIGNS AND SYMPTOMS amyloidosis, FMF patients have normal life expectancy.
Familial Mediterranean Fever Cryopyrin-Associated Periodic Syndrome
FMF is an autosomal recessive disease. Over 90% of patients CAPS is autosomal dominantly inherited. Originally, three separate
become symptomatic within the first two decades of life. Typically, clinical syndromes, all with their own clinical features, were
attacks are characterized by abrupt onset of high fever, peaking distinguished: familial cold autoinflammatory syndrome (FCAS),
soon after onset and lasting for 12 hours to 3 days. Subsequently, Muckle Wells syndrome (MWS), and neonatal onset multisystem
the fever subsides rapidly. Painful serositis accompanies the fever. inflammatory disease (NOMID), which is also known as chronic
Serositis can also be present without fever. Over 95% of patients infantile neurological, cutaneous and articular (CINCA) syn-
experience abdominal pain, which lasts up to 3 days. The pain, drome. With the discovery of NLRP3 mutations in all three
which is caused by sterile peritonitis, may initially be focal and diseases, it has become clear that the clinical phenotype of CAPS
progress to more diffuse pain. Before being diagnosed with FMF, is a continuous spectrum of severity, instead of distinct diseases.
a significant proportion of patients will have undergone explor- There is no genotype–phenotype association, suggesting a role
atory abdominal surgery under suspicion of appendicitis. At for other yet undiscovered disease modifying factors.
surgery, intraabdominal adhesions, a result of recurrent peritonitis, CAPS often manifests clinically soon after birth or in early
may be found. Pelvic adhesions can reduce fertility in female childhood. It is characterized by recurrent urticaria-like rash,
patients. Pleuritis, presenting as thoracic pain, is experienced by arthralgia, myalgia, headache, and fever. Ocular symptoms, in-
approximately 40% of patients. Synovitis with monoarthritis of cluding conjunctivitis and uveitis, are common. Some patients
knee, ankle, or wrist occurs in one-half to three-quarters of develop sensorineural hearing loss during adolescence or adult-
patients. An arthritic attack may have a more protracted course hood. At the severe end of the clinical spectrum, central nervous
compared with nonarthritic FMF, with fever lasting up to a week. system (CNS) symptoms, including chronic aseptic meningitis
Joint pain may persist when fever has already subsided. Synovitis that is characterized by chronic headache, increased intracranial
usually resolves completely without joint destruction. The skin pressure, hydrocephalus, mental retardation, and seizures, are
can be affected. Erysipelas-like skin lesions overlying the shins common. Papilledema with optic nerve atrophy can lead to loss