
Page 227 - The Netter Collection of Medical Illustrations - Integumentary System_ Volume 4 ( PDFDrive )
P. 227
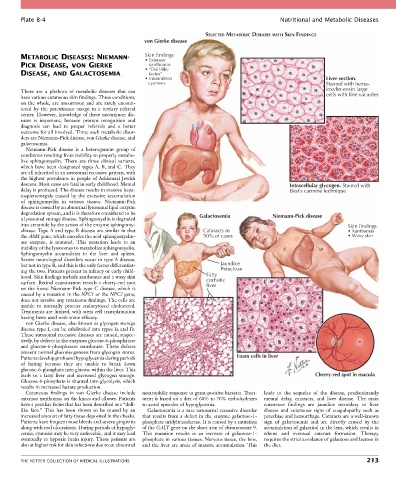
Plate 8-4 Nutritional and Metabolic Diseases
SELECTED METABOLIC DISEASES WITH SKIN FINDINGS
von Gierke disease
METABOLIC DISEASES: NIEMANN- Skin findings
• Extensor
PICK DISEASE, vON GIERKE xanthomas
• “Doll-like
DISEASE, AND GALACTOSEMIA facies”
• Intermittent Liver section.
cyanosis Stained with hema-
There are a plethora of metabolic diseases that can toxylin-eosin large
have various cutaneous skin findings. These conditions, cells with fine vacuoles
on the whole, are uncommon and are rarely encoun-
tered by the practitioner except in a tertiary referral
center. However, knowledge of these uncommon dis-
eases is important, because prompt recognition and
diagnosis can lead to proper referrals and a better
outcome for all involved. Three such metabolic disor-
ders are Niemann-Pick disease, von Gierke disease, and
galactosemia.
Niemann-Pick disease is a heterogenous group of
conditions resulting from inability to properly metabo-
lize sphingomyelin. There are three clinical variants,
which have been designated types A, B, and C. They
are all inherited in an autosomal recessive pattern, with
the highest prevalence in people of Ashkenazi Jewish
descent. Most cases are fatal in early childhood. Mental Intracellular glycogen. Stained with
delay is profound. The disease results in massive hepa- Best’s carmine technique
tosplenomegaly caused by the excessive accumulation
of sphingomyelin in various tissues. Niemann-Pick
disease is caused by an abnormal lysosomal lipid enzyme
degradation system, and it is therefore considered to be Galactosemia Niemann-Pick disease
a lysosomal storage disease. Sphingomyelin is degraded
into ceramide by the action of the enzyme sphingomy- Skin findings
elinase. Type A and type B disease are similar in that Cataracts in • Xanthomas
the ASM gene, which encodes the acid sphingomyelin- 50% of cases • Waxy skin
ase enzyme, is mutated. This mutation leads to an
inability of the lysosomes to metabolize sphingomyelin.
Sphingomyelin accumulates in the liver and spleen.
Severe neurological disorders occur in type A disease,
but not in type B, and this is the only factor differentiat- Jaundice
ing the two. Patients present in infancy or early child- Petechiae
hood. Skin findings include xanthomas and a waxy skin Fatty
surface. Retinal examination reveals a cherry-red spot cirrhotic
liver
on the fovea. Niemann-Pick type C disease, which is
caused by a mutation in the NPC1 or the NPC2 gene,
does not involve any cutaneous findings. The cells are
unable to normally process endocytosed cholesterol.
Treatments are limited, with stem cell transplantation
having been used with some efficacy.
von Gierke disease, also known as glycogen storage
disease type I, can be subdivided into types Ia and Ib.
These autosomal recessive diseases are caused, respec-
tively, by defects in the enzymes glucose-6-phosphatase
and glucose-6-phosphatase translocase. These defects
prevent normal gluconeogenesis from glycogen stores.
Patients develop profound hypoglycemia during periods Foam cells in liver
of fasting because they are unable to break down
glucose-6-phosphate into glucose within the liver. This
leads to a fatty liver and increased glycogen storage. Cherry-red spot in macula
Glucose-6-phosphate is shunted into glycolysis, which
results in increased lactate production.
Cutaneous findings in von Gierke disease include neutrophilic response to gram-positive bacteria. Treat- leads to the sequelae of the disease, predominantly
extensor xanthomas on the knees and elbows. Patients ment is based on a diet of 60% to 70% carbohydrates mental delay, cataracts, and liver disease. The main
have a peculiar facies that has been described as a “doll- to avoid episodes of hypoglycemia. cutaneous findings are jaundice secondary to liver
like face.” This has been shown to be caused by an Galactosemia is a rare autosomal recessive disorder disease and cutaneous signs of coagulopathy such as
increased amount of fatty tissue deposited in the cheeks. that results from a defect in the enzyme galactose-1- petechiae and hemorrhage. Cataracts are a well-known
Patients have frequent nose bleeds and severe gingivitis phosphate uridyltransferase. It is caused by a mutation sign of galactosemia and are directly caused by the
along with oral ulcerations. During periods of hypogly- of the GALT gene on the short arm of chromosome 9. accumulation of galactitol in the lens, which results in
cemia, cyanosis may be very noticeable, and it may lead This mutation results in an increase of galactose-1- edema and eventual cataract formation. Therapy
eventually to hypoxic brain injury. These patients are phosphate in various tissues. Nervous tissue, the lens, requires the strict avoidance of galactose and lactose in
also at higher risk for skin infections due to an abnormal and the liver are areas of massive accumulation. This the diet.
THE NETTER COLLECTION OF MEDICAL ILLUSTRATIONS 213