
Page 759 - Williams Hematology ( PDFDrive )
P. 759
734 Part VI: The Erythrocyte Chapter 48: The Thalassemias: Disorders of Globin Synthesis 735
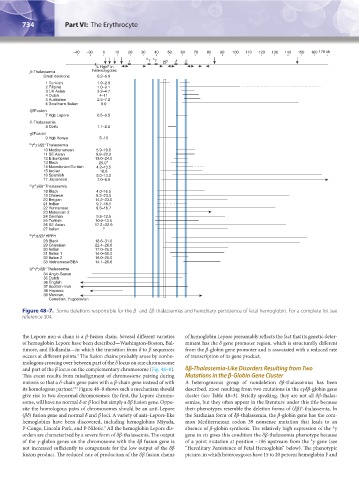
Figure 48–7. Some deletions responsible for the β- and δβ-thalassemias and hereditary persistence of fetal hemoglobin. For a complete list see
reference 304.
the Lepore non-α chain is a β-fusion chain. Several different varieties of hemoglobin Lepore presumably reflects the fact that its genetic deter-
of hemoglobin Lepore have been described—Washington-Boston, Bal- minant has the δ gene promoter region, which is structurally different
timore, and Hollandia—in which the transition from δ to β sequences from the β-globin gene promoter and is associated with a reduced rate
occurs at different points. The fusion chains probably arose by nonho- of transcription of its gene product.
7
mologous crossing over between part of the δ locus on one chromosome
and part of the β locus on the complementary chromosome (Fig. 48–8). δβ-Thalassemia-Like Disorders Resulting from Two
This event results from misalignment of chromosome pairing during Mutations in the β-Globin Gene Cluster
meiosis so that a δ-chain gene pairs with a β-chain gene instead of with A heterogeneous group of nondeletion δβ-thalassemias has been
its homologous partner. Figure 48–8 shows such a mechanism should described, most resulting from two mutations in the εγδβ-globin gene
103
give rise to two abnormal chromosomes: the first, the Lepore chromo- cluster (see Table 48–3). Strictly speaking, they are not all δβ-thalas-
some, will have no normal δ or β loci but simply a δβ fusion gene. Oppo- semias, but they often appear in the literature under this title because
site the homologous pairs of chromosomes should be an anti-Lepore their phenotypes resemble the deletion forms of (δβ) -thalassemia. In
0
(βδ) fusion gene and normal δ and β loci. A variety of anti–Lepore-like the Sardinian form of δβ-thalassemia, the β-globin gene has the com-
hemoglobins have been discovered, including hemoglobins Miyada, mon Mediterranean codon 39 nonsense mutation that leads to an
P-Congo, Lincoln Park, and P-Nilotic. All the hemoglobin Lepore dis- absence of β-globin synthesis. The relatively high expression of the γ
A
7
orders are characterized by a severe form of δβ-thalassemia. The output gene in cis gives this condition the δβ-thalassemia phenotype because
of the γ-globin genes on the chromosome with the δβ fusion gene is of a point mutation at position –196 upstream from the γ gene (see
A
not increased sufficiently to compensate for the low output of the δβ “Hereditary Persistence of Fetal Hemoglobin” below). The phenotypic
fusion product. The reduced rate of production of the δβ fusion chains picture, in which heterozygotes have 15 to 20 percent hemoglobin F and
Kaushansky_chapter 48_p0725-0758.indd 734 9/18/15 2:57 PM