
Page 1005 - Clinical Immunology_ Principles and Practice ( PDFDrive )
P. 1005
CHaPter 72 Immunological Lung Diseases 969
A B
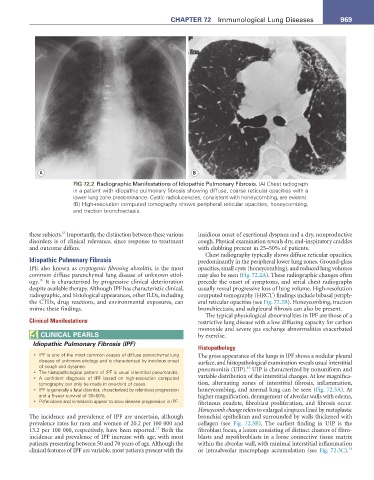
FIG 72.2 Radiographic Manifestations of Idiopathic Pulmonary Fibrosis. (A) Chest radiograph
in a patient with idiopathic pulmonary fibrosis showing diffuse, coarse reticular opacities with a
lower lung zone predominance. Cystic radiolucencies, consistent with honeycombing, are evident.
(B) High-resolution computed tomography shows peripheral reticular opacities, honeycombing,
and traction bronchiectasis.
15
these subjects. Importantly, the distinction between these various insidious onset of exertional dyspnea and a dry, nonproductive
disorders is of clinical relevance, since response to treatment cough. Physical examination reveals dry, end-inspiratory crackles
and outcome differs. with clubbing present in 25–50% of patients.
Chest radiography typically shows diffuse reticular opacities,
Idiopathic Pulmonary Fibrosis predominantly in the peripheral lower lung zones. Ground-glass
IPF, also known as cryptogenic fibrosing alveolitis, is the most opacities, small cysts (honeycombing), and reduced lung volumes
common diffuse parenchymal lung disease of unknown etiol- may also be seen (Fig. 72.2A). These radiographic changes often
16
ogy. It is characterized by progressive clinical deterioration precede the onset of symptoms, and serial chest radiographs
despite available therapy. Although IPF has characteristic clinical, usually reveal progressive loss of lung volume. High-resolution
radiographic, and histological appearances, other ILDs, including computed tomography (HRCT) findings include bibasal periph-
the CTDs, drug reactions, and environmental exposures, can eral reticular opacities (see Fig. 72.2B). Honeycombing, traction
mimic these findings. bronchiectasis, and subpleural fibrosis can also be present.
The typical physiological abnormalities in IPF are those of a
Clinical Manifestations restrictive lung disease with a low diffusing capacity for carbon
monoxide and severe gas exchange abnormalities exacerbated
CLInICaL PearLS by exercise.
Idiopathic Pulmonary Fibrosis (IPF)
Histopathology
• IPF is one of the most common causes of diffuse parenchymal lung The gross appearance of the lungs in IPF shows a nodular pleural
disease of unknown etiology and is characterized by insidious onset surface, and histopathological examination reveals usual interstitial
of cough and dyspnea. 18
• The histopathological pattern of IPF is usual interstitial pneumonitis. pneumonitis (UIP). UIP is characterized by nonuniform and
• A confident diagnosis of IPF based on high-resolution computed variable distribution of the interstitial changes. At low magnifica-
tomography can only be made in one-third of cases. tion, alternating zones of interstitial fibrosis, inflammation,
• IPF is generally a fatal disorder, characterized by relentless progression honeycombing, and normal lung can be seen (Fig. 72.3A). At
and a 5-year survival of 30–50%. higher magnification, derangement of alveolar walls with edema,
• Pirfenidone and nintedanib appear to slow disease progression in IPF. fibrinous exudate, fibroblast proliferation, and fibrosis occur.
Honeycomb change refers to enlarged airspaces lined by metaplastic
The incidence and prevalence of IPF are uncertain, although bronchial epithelium and surrounded by walls thickened with
prevalence rates for men and women of 20.2 per 100 000 and collagen (see Fig. 72.3B). The earliest finding in UIP is the
17
13.2 per 100 000, respectively, have been reported. Both the fibroblast focus, a lesion consisting of distinct clusters of fibro-
incidence and prevalence of IPF increase with age, with most blasts and myofibroblasts in a loose connective tissue matrix
patients presenting between 50 and 70 years of age. Although the within the alveolar wall, with minimal interstitial inflammation
18
clinical features of IPF are variable, most patients present with the or intraalveolar macrophage accumulation (see Fig. 72.3C).