
Page 412 - Textbook of Pathology, 6th Edition
P. 412
396 In man, two of the major risk factors which act together
to produce endothelial injury are: haemodynamic stress from
hypertension and chronic dyslipidaemia. The role of
haemodynamic forces in causing endothelial injury is further
supported by the distribution of atheromatous plaques at
points of bifurcation or branching of blood vessels which are
under greatest shear stress.
ii) Intimal smooth muscle cell proliferation. Endothelial
injury causes adherence, aggregation and platelet release
reaction at the site of exposed subendothelial connective
tissue and infiltration by inflammatory cells. Proliferation of
intimal smooth muscle cell and production of extracellular
matrix are stimulated by various cytokines such as IL-1 and
TNF-α released from invading monocyte-macrophages and
by activated platelets at the site of endothelial injury. These
cytokines lead to local synthesis of following growth factors
having distinct roles in plaque evolution:
Platelet-derived growth factor (PDGF) and fibroblast
growth factor (FGF) stimulate proliferation and migration
of smooth muscle cells from their usual location in the media
into the intima.
Transforming growth factor-β (TGF-β) and interferon-
(IFN-γ) derived from activated T lymphocytes within lesions
regulate the synthesis of collagen by smooth muscle cells.
Smooth muscle cell proliferation is also facilitated by
biomolecules such as nitric oxide and endothelin released from
endothelial cells. Intimal proliferation of smooth muscle cells
is accompanied by synthesis of matrix proteins—collagen,
SECTION III
elastic fibre proteins and proteoglycans.
iii) Role of blood monocytes. Though blood monocytes do
not possess receptors for normal LDL, LDL does appear in
the monocyte cytoplasm to form foam cell by mechanism
illustrated in Fig. 15.5. Plasma LDL on entry into the intima
undergoes oxidation. The ‘oxidised LDL’ formed in the
intima performs the following all-important functions on
monocytes and endothelium:
For monocytes: Oxidised LDL acts to attract, proliferate,
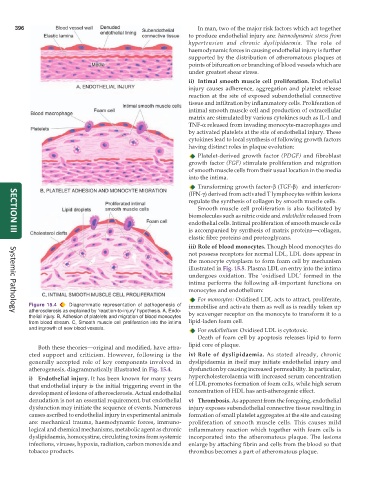
Figure 15.4 Diagrammatic representation of pathogenesis of immobilise and activate them as well as is readily taken up
atherosclerosis as explained by ‘reaction-to-injury’ hypothesis. A, Endo- by scavenger receptor on the monocyte to transform it to a
Systemic Pathology
thelial injury. B, Adhesion of platelets and migration of blood monocytes
from blood stream. C, Smooth muscle cell proliferation into the intima lipid-laden foam cell.
and ingrowth of new blood vessels. For endothelium: Oxidised LDL is cytotoxic.
Death of foam cell by apoptosis releases lipid to form
Both these theories—original and modified, have attra- lipid core of plaque.
cted support and criticism. However, following is the iv) Role of dyslipidaemia. As stated already, chronic
generally accepted role of key components involved in dyslipidaemia in itself may initiate endothelial injury and
atherogenesis, diagrammatically illustrated in Fig. 15.4. dysfunction by causing increased permeability. In particular,
i) Endothelial injury. It has been known for many years hypercholesterolaemia with increased serum concentration
that endothelial injury is the initial triggering event in the of LDL promotes formation of foam cells, while high serum
development of lesions of atherosclerosis. Actual endothelial concentration of HDL has anti-atherogenic effect.
denudation is not an essential requirement, but endothelial v) Thrombosis. As apparent from the foregoing, endothelial
dysfunction may initiate the sequence of events. Numerous injury exposes subendothelial connective tissue resulting in
causes ascribed to endothelial injury in experimental animals formation of small platelet aggregates at the site and causing
are: mechanical trauma, haemodynamic forces, immuno- proliferation of smooth muscle cells. This causes mild
logical and chemical mechanisms, metabolic agent as chronic inflammatory reaction which together with foam cells is
dyslipidaemia, homocystine, circulating toxins from systemic incorporated into the atheromatous plaque. The lesions
infections, viruses, hypoxia, radiation, carbon monoxide and enlarge by attaching fibrin and cells from the blood so that
tobacco products. thrombus becomes a part of atheromatous plaque.