
Page 255 - Williams Hematology ( PDFDrive )
P. 255
230 Part IV: Molecular and Cellular Hematology Chapter 16: Cell-Cycle Regulation and Hematologic Disorders 231
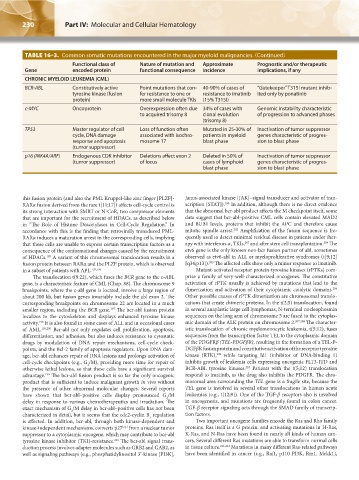
TABLE 16–3. Common somatic mutations encountered in the major myeloid malignancies. (Continued)
Functional class of Nature of mutation and Approximate Prognostic and/or therapeutic
Gene encoded protein functional consequence incidence implications, if any
CHRONIC MYELOID LEUKEMIA (CML)
BCR-ABL Constitutively active Point mutations that con- 40-90% of cases of “Gatekeeper” T315I mutant inhib-
tyrosine kinase (fusion fer resistance to one or resistance to imatinib ited only by ponatinib
protein) more small molecule TKIs (15% T315I)
c-MYC Oncoprotein Overexpression often due 34% of cases with Genomic instability characteristic
to acquired trisomy 8 clonal evolution of progression to advanced phases
(trisomy 8)
TP53 Master regulator of cell Loss of function often Mutated in 25-30% of Inactivation of tumor suppressor
cycle, DNA damage associated with isochro- patients in myeloid genes characteristic of progres-
response and apoptosis mosome 17 blast phase sion to blast phase
(tumor suppressor)
p16 (INK4A/ARF) Endogenous CDK inhibitor Deletions affect exon 2 Deleted in 50% of Inactivation of tumor suppressor
(tumor suppressor) of locus cases of lymphoid genes characteristic of progres-
blast phase sion to blast phase
this fusion protein (and also the PML Kruppel-like zinc finger [PLZF]- Janus-associated kinase [JAK]–signal transducer and activator of tran-
198
RARα fusion derived from the rare t[11;17]) affects cell-cycle control is scription [STAT]). In addition, although there is no direct evidence
its strong interaction with SMRT or N-CoR, two corepressor elements that the abnormal bcr-abl product affects the M checkpoint itself, some
that are important for the recruitment of HDACs, as described below data suggest that bcr-abl–positive CML cells contain elevated MAD2
in “The Role of Histone Deacetylases in Cell-Cycle Regulation.” In and BUB1 levels, proteins that inhibit the APC and therefore cause
202
accordance with this is the finding that retrovirally transduced PML- mitotic spindle arrest. Amplification of the fusion sequence is fre-
RARα induces a maturation arrest in the corresponding cells, implying quently used to detect minimal residual disease in patients under ther-
that these cells are unable to express certain transcription factors as a apy with interferon-α, TKIs, and after stem cell transplantation. The
203
204
consequence of the conformational changes caused by the recruitment etv6 gene is the only known non-bcr fusion partner of abl, sometimes
of HDACs. A variant of this chromosomal translocation results in a observed as etv6-abl in ALL or myeloproliferative syndromes (t[9;12]
195
205
fusion protein between RARα and the PLZF protein, which is observed [q34;p13]). The affected cells show only a minor response to imatinib.
in a subset of patients with APL. 195,196 Mutant-activated receptor protein-tyrosine kinases (rPTKs) com-
The translocation t(9:22), which fuses the BCR gene to the c-ABL prise a family of very-well-characterized oncogenes. The constitutive
gene, is a characteristic feature of CML (Chap. 88). The chromosome 9 activation of rPTK usually is achieved by mutations that lead to the
breakpoints, where the c-abl gene is located, involve a large region of dimerization and activation of their cytoplasmic catalytic domains.
206
about 200 kb, but fusion genes invariably include the abl exon 2. The Other possible causes of rPTK dimerization are chromosomal translo-
corresponding breakpoints on chromosome 22 are located in a much cations that create chimeric proteins. In the t(2;5) translocation, found
smaller region, including the BCR gene. The bcr-abl fusion protein in several anaplastic large cell lymphomas, N-terminal nucleophosmin
197
localizes to the cytoskeleton and displays enhanced tyrosine kinase sequences on the long arm of chromosome 5 are fused to the cytoplas-
activity. It is also found in some cases of ALL and in occasional cases mic domain of the ALK protein on chromosome 2. 207,208 The character-
198
of AML. 199,200 Bcr-abl not only regulates cell proliferation, apoptosis, istic translocation of chronic myelomonocytic leukemia, t(5;12), fuses
differentiation, and adhesion, but also induces resistance to cytostatic sequences from the transcription factor TEL to the cytoplasmic domain
drugs by modulation of DNA repair mechanisms, cell-cycle check- of the PDGFRβ (TEL-PDGFβR), resulting in the formation of a TEL-P-
points, and the Bcl-2 family of apoptosis regulators. Upon DNA dam- DGFβR fusion protein and constitutive activation of the receptor tyrosine
209
age, bcr-abl enhances repair of DNA lesions and prolongs activation of kinase (RTK), while targeting Id1 (inhibitor of DNA-binding 1)
cell-cycle checkpoints (e.g., G /M), providing more time for repair of inhibits growth of leukemia cells expressing oncogenic FLT3-ITD and
2
otherwise lethal lesions, so that these cells have a significant survival BCR-ABL tyrosine kinases. Patients with the t(5;12) translocation
210
advantage. The bcr-abl fusion product is so far the only oncogenic respond to imatinib, as the drug also inhibits the PDGFR. The chro-
199
product that is sufficient to induce malignant growth in vivo without mosomal area surrounding the TEL gene is a fragile site, because the
the presence of other abnormal molecular changes. Several reports TEL gene is involved in several other translocations in human acute
have shown that bcr-abl–positive cells display pronounced G /M leukemias (e.g., t[12;9]). One of the TGF-β receptors also is involved
2
delay in response to various chemotherapeutics and irradiation. The in oncogenesis, and mutations are frequently found in colon cancer.
exact mechanism of G /M delay in bcr-abl–positive cells has not been TGF-β receptor signaling acts through the SMAD family of transcrip-
2
characterized in detail, but it seems that the cdc2-cyclin B regulation tion factors.
1
is affected. In addition, bcr-abl, through both kinase-dependent and Two important oncogene families encode the Ras and Rho family
kinase-independent mechanisms, converts p27 kip1 from a nuclear tumor proteins. Ras itself is a G protein, and activating mutations in H-Ras,
suppressor to a cytoplasmic oncogene, which may contribute to bcr-abl K-Ras, and N-Ras have been found in nearly all kinds of human can-
tyrosine kinase inhibitor (TKI)-resistance. The bcr-abl signal trans- cers. Several different Ras mutations are able to transform normal cells
201
duction process involves adapter molecules such as GRB2 and GAB2, as in tissue culture. 211,212 Mutations in many different Ras-related pathways
well as signaling pathways (e.g., phosphatidylinositol 3′-kinase [PI3K], have been identified in cancer (e.g., Raf1, p110 PI3K, Rin1, Mekk1),
Kaushansky_chapter 16_p0213-0246.indd 230 9/18/15 11:57 PM