
Page 339 - Textbook of Pathology, 6th Edition
P. 339
β β β β β-Thalassaemia Major 323
β-thalassaemia major, also termed Mediterranean or Cooley’s
anaemia is the most common form of congenital haemolytic
anaemia. β-thalassaemia major is a homozygous state with
either complete absence of β-chain synthesis (β° thalassaemia
+
major) or only small amounts of β-chains are formed (β thalas-
saemia major). These result in excessive formation of alternate
haemoglobins, HbF (α γ ) and HbA (α δ ).
2
2 2
2
2
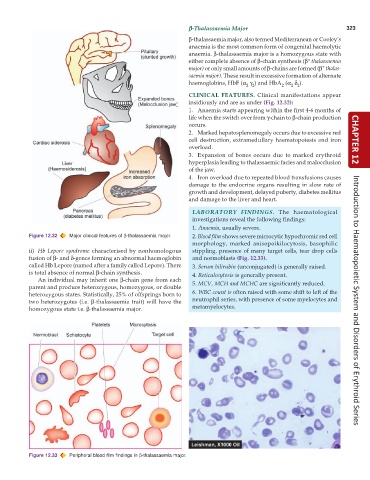
CLINICAL FEATURES. Clinical manifestations appear
insidiously and are as under (Fig. 12.32):
1. Anaemia starts appearing within the first 4-6 months of
life when the switch over from γ-chain to β-chain production
occurs.
2. Marked hepatosplenomegaly occurs due to excessive red
cell destruction, extramedullary haematopoiesis and iron CHAPTER 12
overload.
3. Expansion of bones occurs due to marked erythroid
hyperplasia leading to thalassaemic facies and malocclusion
of the jaw.
4. Iron overload due to repeated blood transfusions causes
damage to the endocrine organs resulting in slow rate of
growth and development, delayed puberty, diabetes mellitus
and damage to the liver and heart.
LABORATORY FINDINGS. The haematological
investigations reveal the following findings:
1. Anaemia, usually severe.
Figure 12.32 Major clinical features of β-thalassaemia major. 2. Blood film shows severe microcytic hypochromic red cell
morphology, marked anisopoikilocytosis, basophilic
ii) Hb Lepore syndrome characterised by nonhomologous stippling, presence of many target cells, tear drop cells
fusion of β- and δ-genes forming an abnormal haemoglobin and normoblasts (Fig. 12.33).
called Hb Lepore (named after a family called Lepore). There 3. Serum bilirubin (unconjugated) is generally raised.
is total absence of normal β-chain synthesis. 4. Reticulocytosis is generally present.
An individual may inherit one β-chain gene from each
parent and produce heterozygous, homozygous, or double 5. MCV, MCH and MCHC are significantly reduced.
heterozygous states. Statistically, 25% of offsprings born to 6. WBC count is often raised with some shift to left of the
two heterozygotes (i.e. β-thalassaemia trait) will have the neutrophil series, with presence of some myelocytes and Introduction to Haematopoietic System and Disorders of Erythroid Series
homozygous state i.e. β-thalassaemia major. metamyelocytes.
Figure 12.33 Peripheral blood film findings in β-thalassaemia major.